Fabienne Ballanger1, Sébastien Barbarot1, Steven Le Gouill2, Fanny Gaillard3, Elisabeth Cassagnau3, Laurence Lodé4, Brigitte Dréno1 and Jean F. Stalder1
Departments of 1Dermatology, 2Haematology, 3Anatomopathology and 4Molecular Biology, Hôtel Dieu, FR-44035 Nantes, France. E-mail: fabienne.ballanger@chu-nantes.fr
Accepted March 12, 2009.
Sir,
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is an uncommon type of peripheral T-cell lymphoma. The classical clinical presentation is erythematous, subcutaneous plaques or nodules involving the extremities and the trunk. Two distinctive entities are described: SPTCL with α/β or γ/δ phenotype. We report here an atypical clinical presentation of SPTCL, illustrating the difficulties of diagnosis of this kind of T-cell lymphoma.
CASE REPORT
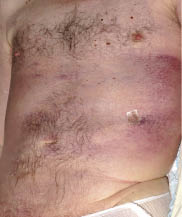
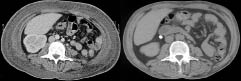
A 54-year-old man, with no medical history, was referred for evaluation of an erythematous, oedematous subcutaneous infiltration of the trunk. The symptoms had begun 2 months earlier with numerous, tender, erythematous nodules of the trunk, associated with fever up to 39°C that persisted despite antibiotics. Very quickly, the cutaneous symptoms changed into an erythematous, oedematous thickening of the skin of the trunk with no visible nodules (Fig. 1). This was associated with a weight gain of 10 kg. No lymphadenopathy was found. Laboratory studies revealed anaemia (haemoglobin: 10 g/dl), elevated liver function test findings (serum glutamate oxaloacetate transaminase: 156 UI/l; serum glutamate pyruvate transaminase: 128 UI/l; normal value < 30 UI/l), elevated lactic dehydrogenase (LDH) 1707 UI/l (normal < 420 UI/l), hyperferritinaemia 13,946 µg/l (normal < 80 µg/l) and hypertriglyceridaemia 5.61 g/l (normal < 1.7g/l). Immunological and bacteriological laboratory studies and viral serologies were all negative. Several punch biopsies were performed. In each of these the epidermis showed reactive changes, with no epidermotropism, associated with a mild dermal inflammatory infiltrate simulating panniculitis. However, the biopsies were too superficial to conclude. A thoracic/abdominal computed tomography (CT) scan showed a diffused subcutaneous thoracic and abdominal wall infiltration, hepatosplenomegaly without deep lymphadenopathy (Fig. 2a). Bone marrow biopsy did not show haemophagocytosis or involvement by lymphoma.
Fig. 1. Clinical presentation of subcutaneous panniculitis-like T-cell lymphoma as diffuse thoracic subcutaneous infiltration.
Fig. 2. Computed tomography scan (left) before and (right) after chemotherapy: subcutaneous infiltration of the trunk and diminution of the subcutaneous infiltration.
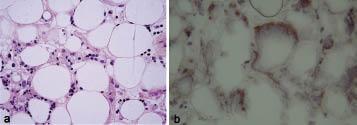
A deep incisional biopsy specimen of the abdominal infiltration was performed. It displayed aggregates of foamy histiocytes and lymphocytes extending into subcutaneous fat associated with necrosis and karryorrhexis (Fig. 3a). The infiltrate was mild and consisted of small, medium and large lymphoid cells with irregular nuclear contour and densely clumped chromatin. These atypical lymphocytes were rimming fat lobules.
Immunophenotypical analysis in paraffin sections showed that the neoplastic cells were of T-cell phenotype, stained with CD45, CD3, and CD8, but CD56- and CD30-negative. In addition, there was CD7 antigen loss. CD68+ staining revealed the presence of histiocytes. Molecular biological studies (polymerase chain reaction (PCR)) of α/β T cell receptor (TCR) in the skin and the blood revealed clonal T-cell population. Immunohistochemistry studies using anti-human α/β TCR and anti-human γ/δ TCR (mouse IgG1k, Ebioscience, Montrouge, France) confirmed that TCR rearrangement was α/β+ (Fig. 3b).
Fig. 3. (a) Foamy histiocytes with karryorrhexis and atypical T cells rimming individual adipocytes (×400). (b) Immunohistochemistry using anti-human α/β T-cell receptor antibody (×400).
Thus, a diagnosis of α/β SPTCL was made. Despite normal bone marrow, clinical and biological symptoms (fever, hepatosplenomegaly, cytopaenia, hyperferritinaemia and hypertryglyceridaemia) suggested an association with haemophagocytic syndrome.
The patient was treated with eight cycles of CHOP chemotherapy (cyclophosphamide (750 mg/m2 day 1), doxorubicin (50 mg/m2 day 1), vincristine (1.4 mg/m2 day 1) and prednisone (80 mg/m2 per day 1–5)) with partial clinical remission. A CT scan performed after eight cycles of CHOP revealed persistent mild subcutaneous infiltration, confirmed by positron emisson tomography (PET) scan, but hepatosplenomegaly had regressed to normal size (Fig. 2b). Biological abnormalities were almost corrected, with only persistent elevated LDH (1.5 N). Due to borderline persistent thrombopaenia (82 × 109/l without sign of relapse: normal bone marrow and negative analysis for T-cell receptor gene rearrangement), CHOP chemotherapy was stopped and autologous stem cell transplantation was proposed, but was refused by the patient. Maintenance chemotherapy with NIPENT (pentostatin 4 mg/m2 every 3 weeks) and dexamethasone was commenced. Twelve cycles were realized, with good biological and clinical tolerance. At the present time, clinical and radiological evaluation shows that the patient has remained in a stable condition. The follow-up duration after obtaining partial remission is 10 months since the end of the chemotherapy.
DISCUSSION
SPTCL is a rare primary cutaneous T-cell lymphoma, first described by Gonzales et al. in 1991 (1). It is currently classified in the category of peripheral T-cell neoplasms (2). The median age at diagnosis is approximately 39 years. The classical clinical presentation consists of tender erythematous nodules, and the lower extremities are the most frequent sites of cutaneous involvement, followed by the trunk, arms and face. Ulceration occurs in 20% of cases (3). Biologically, cytopaenia occurs in approximately 20% of cases and liver dysfunction, as presented by our patient, occurs in 10%. Histologically, SPTCL is characterized by lobular or mixed septal and lobular panniculitis, often combined with cytophagocytosis. Malignant lymphocytes are small to medium size with cytological atypia, which may be minimal, as in our case. This explains why it is frequently misdiagnosed as a benign inflammatory disorder. Typically, lymphocytes infiltrate the fat in a lobular lace-like pattern, rimming the fat spaces, and are associated with karryorrhexis. Immunohistochemical staining is positive for T-cell antigen. CD7 antigen loss is observed in 30% of patients (4). Clonal rearrangement of TCR, shown by PCR, confirms T-cell origin and clonality in 85% of cases.
In recent years, it has been observed that although SPTCL with α/β phenotype has an excellent prognosis, in particular when not complicated by haemophagocytic syndrome (HPS), a rapid clinical deterioration secondary to HPS is frequent in SPTCL with γ/δ phenotype (3). HPS is a multisystem illness characterized by fever, wasting, adenopathy, hepatosplenomegaly, pancytopaenia, coagulopathy, hyperferritinaemia and hypertriglyceridaemia. It develops in 45–70% of patients with SPTCL, with a mortality rate of 81% (4, 5). HPS associated with panniculitis has even been reported without any clear evidence of malignant lymphoma (4, 6). HPS, cytopaenia, and elevated LDH appear to be poor prognostic factors (7–9).
The standard treatment of the disease has not yet been established. As SPTCL with α/β phenotype and SPTCL with γ/δ phenotype appear to be two distinct entities with different prognoses, treatment should be adapted (2). Patients presenting with α/β TCR+SPTCL without HPS may benefit equally either from prednisone or others immunosuppressive agents. In the case of a solitary lesion, radiotherapy may be considered. However, low-dose chemotherapeutic regimens (e.g. CHOP), sometimes associated with local radiotherapy, may be efficient (10). In the case of γ/δ SPTCL, early initiation of aggressive chemotherapy followed by stem-cell transplantation is recommended for better clinical outcome (2). In one case, cyclosporine has been of benefit in SCPTL refractory to CHOP (11). More recently, Hathaway et al. (12) have reported the efficiency of denileukin diftitox treatment in association with corticosteroids (12). In our case, due to the presence of HPS, it was rapidly decided to use multi-agent chemotherapy. This could explain the partial clinical response even though the patient presented with few poor prognostic factors. However, stem-cell transplantation was refused by our patient and pentostatin was chosen for maintenance treatment. Pentostatin is a potent inhibitor of adenosine deaminase and is selectively toxic to lymphocytes. It has been used in human γ/δ+ T-cell malignancies (13) and in cutaneous T-cell lymphoma, especially Sézary syndrome (14, 15).
REFERENCES