Suhyun Cho1, Kyung Jong Lee2, Jong Doo Lee3, Dongsik Bang1 and Sung Bin Cho1*
1Department of Dermatology and Cutaneous Biology Research Institute, 2Internal Medicine, 3Division of Nuclear Medicine, Department of Diagnostic Radiology, Yonsei University College of Medicine, 250 Seongsanno, Seodaemun-gu, Seoul, Korea. *E-mail: sbcho@yuhs.ac
Accepted November 24, 2010.
Behçet’s disease (BD) is a chronic, multisystemic vasculitis with an unidentified aetiopathogenesis, but there are possible environmental triggering factors such as infectious agents, which have long been postulated as an aetiological factor, including the Herpes simplex virus, Streptococcus sanguis, and Mycobacterium tuberculosis.
CASE REPORT
A 38-year-old woman presented with oral ulcers, genital ulcers, and erythema nodosum-like skin lesions on the legs that had emerged one month previously. Upon physical examination, multiple minor ulcerations were seen on the oral mucosa and vulva, with multiple erythematous, tender nodules, and patches on both lower legs. She also presented with arthralgia, weight loss, and general weakness. No eye problems were detected upon ophthalmological examination. She had had a BCG vaccination but did not have any other medical or family history, including tuberculosis infection. During her initial visit, chest radiography and laboratory tests were performed, including the following: complete blood count, blood glucose level, renal and liver function, erythrocyte sedimentation rate (ESR), C-reactive protein, anti-streptolysin O titre, rheumatoid factor, anti-cyclic citrullinated peptide antibodies, antinuclear antibodies, venereal disease, and HLA B51 genotype. Results were unremarkable, with the exception of an elevated ESR of 74 mm/h (normal range ≤ 20 mm/h) and positive HLA B51 genotype. She was diagnosed with an incomplete type of BD, based on the International Study Group for BD and the revised criteria of the BD Research Committee of Japan (1, 2). She was treated with systemic colchicine, prednisolone, pentoxifylline, and antibiotics for 2 months. However, her symptoms were poorly controlled, with short remissions and recurrent exacerbations in spite of additional administration of daily azathioprine.
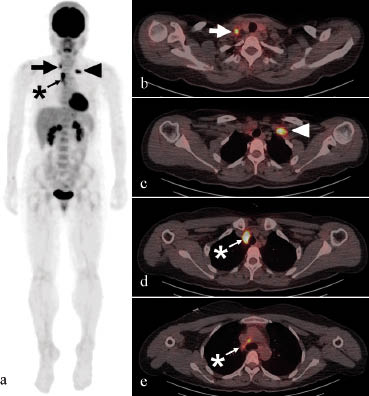
After obtaining informed consent, whole-body 18F-fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) was performed to exclude the possibility of large vessel vasculitis and associated occult malignancies, including lymphoma, because the patient complained of weight loss and persistent general weakness as well as chest discomfort and night sweating following immunosuppressive therapy. Multifocal FDG uptake was detected in the bilateral supraclavicular and right upper paratracheal lymph nodes (Fig. 1). No remarkable uptake was observed in the oropharynx, genital area, subcutaneous tissues, large vessels, or ileocecal area of the intestine. For the evaluation of FDG-positive lymph nodes, ultrasonography-guided lymph node aspiration cytology was performed, but no evidence of malignancy was found. Routine fibreoptic bronchoscopy revealed no endobronchial lesions, and endobronchial ultrasonography-guided lymph node aspiration cytology showed only a few scattered lymphocytes. However, a lymph node biopsy showed chronic granulomatous inflammation with necrosis in several samples, with Ziehl-Neelsen staining revealing a few acid-fast bacilli. Therefore, she was diagnosed with tuberculous lymphadenopathy. The results of an interferon-γ release assay using QuantiFERON tuberculosis Gold (QFT-G; Cellestis Inc, Valencia, CA, USA) were also positive, and quadruple anti-tuberculous medication, comprised of rifampicin, isoniazid, ethambutol, and pyrazinamide, was started. After a month of tuberculosis medication, her weight loss, night sweating, persistent general weakness, orogenital ulcerations, and erythema nodosum-like skin lesions were much improved. However, because the patient presented recurrent, but not severe, oral ulcerations and arthritis, systemic treatment with colchicine and rebamipide had been maintained.
Fig. 1. Whole-body 18F-fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) scan showing multifocal areas of increased FDG uptake: (a) a coronal view of the three-dimensional reconstructed image and (b–e) horizontal views of the PET/CT scan in the right (arrows) and left (arrowheads) supraclavicular lymph nodes and paratracheal lymph nodes (asterisks).
DISCUSSION
BD is a chronic, multisystemic vasculitis that mainly affects small blood vessels (3). Although the exact aetiopathogenesis of BD has not yet been identified, the effects of possible environmental triggering factors, especially infectious agents, in patients with a background of genetic susceptibility are widely accepted as a mainstay of the pathogenesis (4). Infectious agents, which have long been postulated as an aetiological factor, include the Herpes simplex virus, Streptococcus sanguis, and M. tuberculosis (3–5).
The association of BD and tuberculosis has also been reported in some cases, but is known to be rare (6–8). The patients, who satisfied the diagnostic criteria for BD (1, 2) and had been treated with systemic steroid and/or thalidomide, subsequently developed pulmonary tuberculosis (6–8). Recurrent orogenital ulcers presented very good clinical response or complete resolution after anti-tuberculosis medication, suggesting a strong association with BD and M. tuberculosis (6–8). Direskeneli et al. (5) demonstrated significantly increased T-cell proliferative responses to 65 kDa mycobacterial and 60 kDa human heat shock protein in Turkish BD patients observed in the UK and Japan. Furthermore, HLA B51 antigen has been a well-known genetic factor, which plays a pivotal role in determining susceptibility of BD (9). Vijaya Lakshmi et al. (10) reported that HLA B51 has a susceptible association for pulmonary tuberculosis, whereas HLA B52 has a protective association.
It is not clear whether tuberculous lymphadenopathy can be regarded as an initiating or aggravating factor in our BD patient. Moreover, the mucocutaneous symptoms of our patient, such as the orogenital ulcerations and erythema nodosum-like skin lesions, could be regarded as linked with occult tuberculosis infection, rather than with BD (11). However, considering that: (i) the patient showed positive HLA B51 genotyping representing genetic susceptibility to BD; (ii) the diagnostic criteria for BD of the International Study Group for BD and the revised criteria of the BD Research Committee of Japan were satisfied (1, 2); and (iii) the clinical improvements, but not disappearance, of BD symptoms were noted after starting tuberculosis medication, we suggest that tuberculous lymphadenopathy and symptoms associated with mycobacterial infection acted as aggravating factors of BD. In our department, tuberculin skin tests are not routinely performed in BD patients without a past medical history of tuberculosis infection presenting a clear chest radiograph and consideration given to the risk of a pathergy reaction. However, we think that the interferon-γ release assay can be helpful in assessing the possibilities of tuberculosis in recalcitrant BD patients with frequent exacerbations. Moreover, the interferon-γ release assay seems to be more appropriate than tuberculin skin test in BD patients with BCG vaccination history or who are undergoing immunosuppressive therapy.
This report demonstrates the detection of tuberculous lymphadenopathy as an aggravating factor by PET/CT in a patient with BD. We suggest that recalcitrant BD patients with frequent exacerbations may need work-up studies to exclude the possibility of large vessel vasculitis and associated occult malignancies or infections, especially tuberculosis.
References