


Takuya Takeichi1,2, Kazumitsu Sugiura1, Hidee Arai3, Ken Ishii4, Michihiro Kono1 and Masashi Akiyama1*
Departments of Dermatology, 1Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, 2Inazawa City Hospital, Inazawa, 3Department of Neurology, Chiba Children’s Hospital, Chiba, and 4Department of Dermatology, Teikyo University Chiba Medical Center, Ichihara, Japan. *E-mail: makiyama@med.nagoya-u.ac.jp
Accepted October 3, 2012.
Sjögren-Larsson syndrome (SLS: MIM# 270200) is an autosomal recessive hereditary disorder characterized by congenital ichthyosis, mental retardation, and spastic di- or tetraplegia (1). De Laurenzi et al. (2) reported that mutations in the fatty aldehyde dehydrogenase (FALDH) gene (ALDH3A2) were responsible for the development of SLS.
In 1972, a combination of associated defects was named using the acronym VATER; the originally described conditions included Vertebral defects, Anal atresia, Tracheo-oEsophageal fistula with oesophageal atresia, and radial and Renal dysplasia (3). The disorder was later expanded to include Cardiac malformations and a less strict definition of Limb anomalies, and was termed VACTERL association (VA) (4).
The present study reports a Japanese case with VA and genetically diagnosed SLS.
Case report
The patient was a 2.5-year-old Japanese boy. He was born at 30 weeks gestational age by caesarean section due to premature rupture of membranes. His birth weight was 1,948 g. There was no family history of consanguinity. His mother had mild atopic dermatitis. Cutaneous examination revealed fine scales and slight erythema over the whole body, and hyperkeratosis was seen on the hands and feet (Fig. 1A–C). His hair, nails and teeth were normal. He showed serious mental retardation. While spastic tetraplegia was unremarkable, he had ankle clonuses, suggesting pyramidal sign. A skin biopsy specimen showed hyperkeratosis with mild hypergranulosis (Fig. 1D). T2-weighted magnetic resonance imaging demonstrated a high-intensity area in the periventricular white matter of his brain. Moreover, he had multiple congenital anomalies. He had a high anal atresia and several renal anomalies, including left renal hypoplasia, vesicoureteric reflux, hypospadia and a neurogenic bladder. Echocardiography demonstrated ventricular septal defect. He had limb asymmetry and talipes valgus. His vertebrae and oesophagus were normal.
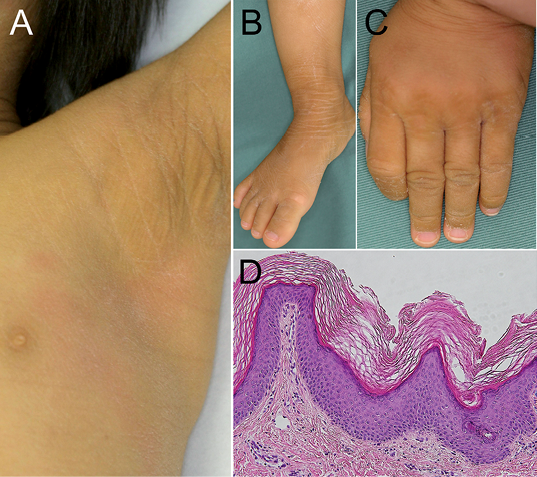
Fig. 1. Clinical features of the patient with Sjögren-Larsson syndrome. At 2 years of age, fine, whitish scales are seen on his (A) armpit, (B) dorsal foot, and (C) hand. (D) Histopathology shows hyperkeratosis with mild hypergranulosis.
The younger sister of the present patient, a 1.5-year-old girl, also had congenital ichthyosis, mental retardation and ankle clonuses on both her lower extremities. Physical examination revealed pigmentation with mild to moderate hyperkeratosis and fine, whitish scales on her neck, trunk, legs and dorsal feet. No other abnormalities were observed.
After written informed consent, FALDH gene (ALDH3A2) mutational analysis was performed on the affected patients and their parents, as previously described (5). In the children, 2 known heterozygous ALDH3A2 mutations, a missense mutation c.1157A>G (p.Asn386Ser) in exon 8 (Fig. S1A; available from http://www.medicaljournals.se/acta/content/?doi=10.2340/00015555-1526) and a deletion mutation c.1291_1292delAA (p.Lys431Glufs*5) in exon 9 (Fig. S1B), were identified. Only missense mutation c.1157A>G was present in the mother, and the deletion mutation c.1291_1292delAA was demonstrated as paternal.
Both mutations have been reported as only homozygous to date (5, 6). The maternal mutation c.1157A>G in exon 8 resulted in the alteration of an asparagine at codon 386 to a serine. FALDH amino-acid sequence alignment shows that this asparagine residue at codon 386 is conserved among diverse species (reference sequences: CR 457422, XP 511337, NP 113919, CAI 25890, NP 001016537, AAK 49120) (Fig. S1C). In addition, the analysis of the secondary structure of the rat class 3 aldehyde dehydrogenase revealed that Asn 388, which corresponds to Asn 386 in human FALDH, appears to stabilize adjacent elements of secondary structure (Fig. S1D) (7). These findings therefore suggest that the substitution of the strictly conserved amino acid asparagine at position 386 could account for the enzyme deficiency of FALDH in patients with SLS. The deletion mutation in exon 9 leads to a frame shift causing a stop codon at 5 codons downstream from the deletion site (p.Lys431Glufs*5). This premature translation termination eliminates the downstream 11% of ALDH3A2 coding sequences.
Discussion
VA is clinically defined by the presence of a cluster of congenital malformations. There is still no firm consensus regarding strict diagnostic criteria, although most clinicians and researchers require the presence of at least 3 component features for diagnosis (8). The present patient had evidence of 4 such features (anal atresia, cardiac malformation, renal anomalies and limb anomalies), and had congenital ichthyosis due to SLS. Solomon et al. (9) reported that a lack of vertebral malformation is unusual in VA. In light of this, the present case might be diagnosed as atypical VA. In some VA patients, other associated anomalies not included in the definition of VA have been detected. Faivre et al. (10) reported that Fanconi’s anaemia patients with VA phenotype had skin pigmentation abnormalities. As far as we know, there is no report of VA patients who had ichthyosis.
Approximately 90% of cases of VA appear to be sporadic, with little increased risk of multiple affected individuals within a family (11). Single or multiple malformations associated with VA are observed in up to 10% of first-degree relatives of patients with VA; in other words, there is evidence for an inherited component in a subset of patients (11). In the present pedigree, although a heterozygous combination of 2 ALDH3A2 mutations has been identified in both children, only the older brother showed the VA phenotype. Thus, we do not consider him to be a genetically inherited case, but rather a sporadic case of VA. However, we cannot exclude the possibility that ALDH3A2 mutations are associated with VA, because the aetiologies of VA remain largely unknown (8, 11). Due to many features of VA, including clinical and causal heterogeneity, the typical sporadic nature of the disease, and the many overlapping conditions, it is difficult to clarify the cause and pathomechanisms of VA (8).
AcknowledgEments
This study was supported in part by a Grant-in-Aid for Scientific Research (A) 23249058 (MA) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by Research on Measures for Intractable Diseases Project: matching fund subsidy (H23-028) from Ministry of Health, Labour and Welfare of Japan.
References


