


Julie Christiansen1, Robin Kahn2, Artur Schmidtchen1 and Karin Berggård1*
1Division of Dermatology and Venereology, and 2Department of Pediatrics, Department of Clinical Sciences Lund, Lund University, SE-221 85 Lund, Sweden. *E-mail: Karin.Berggard@skane.se
Accepted Jun 5, 2014; Epub ahead of print Jun 10, 2014
Angioedema is a potentially life-threatening disease categorised into several forms such as allergic, hereditary, acquired autoimmune and idiopathic. Allergic angioedema is dependent on histamine release, whereas mutations in the C1-inhibitor and factor XII genes have been described in hereditary angioedema (1, 2). Bradykinin-induced acquired angioedema is often caused by antibodies to C1-inhibitor, increased consumption of C1-inhibitor in cases of malignancies or autoimmune diseases (2, 3). Angioedema may also be idiopathic (4). Bradykinin, released during activation of the kallikrein-kinin system, is the main mediator of oedema in hereditary angioedema. It is assumed that the kallikrein-kinin system, linked to the coagulation system via factor XI and factor XII, is also activated in cases of idiopathic angioedema (5, 6). Here, we report an unusual case of therapy resistant idiopathic angioedema, normocomplementaemic urticarial vasculitis and a history of autoimmune acquired haemophilia. Emerging evidence suggests that coagulation and inflammatory responses are interlinked (7–9), as illustrated by this case.
CASE REPORT
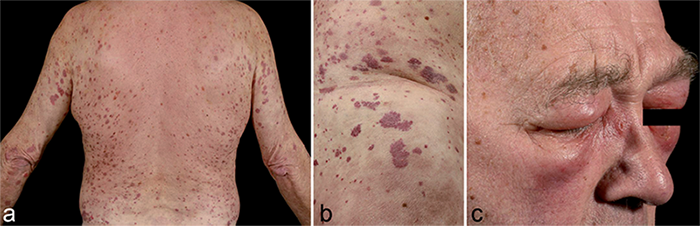
A 71-year-old man was, apart from a history of hypertension and a minor stroke without sequelae, healthy until he developed a peri-/myocarditis of unknown cause. One year later he developed autoimmune haemophilia with antibodies to factor VIII (10). The patient had no other autoimmune disease at the time of diagnosis, but had a high titre of anti-nuclear antibodies (ANA; ≥ 54 IU/ml), speckled pattern. Following treatment with high-dose glucocorticoids, anti-factor VIII autoantibodies could no longer be detected. However, while the patient was still on prednisolone 10 mg/day (less than 5 months after the diagnosis of autoimmune haemophilia), bruises were gradually replaced by non-pruritic erythematous urticarial lesions that remained for more than 24 h and healed with hyperpigmentation (Fig. 1a and b). Skin biopsies showed leucocytoclastic vasculitis and the patient was diagnosed with urticarial vasculitis. Immunofluorescence analysis showed fibrinogen deposits around blood vessels. Subsequently, severe episodes of angioedema (Fig. 1c), including the face, extremities and upper airways, occurred on multiple occasions and the patient underwent emergency coniotomy on one occasion. Angioedema and urticarial vasculitis always occurred concurrently. Treatment with antihistamines, hydroxychloroquine, tranexamic acid or prednisolone (≤ 80 mg/day) had no effect.
A combination of azathioprine 100 mg and dapsone 150 mg daily finally rendered complete recovery from skin lesions and angioedema, but due to side effects, dapsone was discontinued after a few weeks. For almost 3 years the patient was successfully treated with azathioprine. About a month after azathioprine was tapered down, urticarial vasculitis and angioedema reappeared and treatment with azathioprine in high dose was insufficient, even in combination with high dose prednisolone. After 4 months the patient was given colchicine in combination with azathioprine, which finally relieved symptoms. The patient is now since almost 4 years on a maintenance treatment of azathioprine and colchicine.
Apart from medications given to treat vasculitis and angioedema, the patient was taking esomeprazole, but no other drugs. He had never been given ACE-inhibitors. There was no family history of angioedema or connective tissue disease and no underlying disease could be found. Levels of C3 and C4 were normal, whereas levels of C1q and C1-inhibitor were elevated (Table SI1). C1-inhibitor function, complement activation, level of ACE, IgE-levels, histamine-release test for chronic urticaria, ENA, ANCA, anti-ds-DNA, anti-C1q and anti-cardiolipin antibodies were all normal. Apart from a high titre of speckled pattern ANA, an APTT in the upper normal range, decreased levels of factor XII and highly elevated levels of bradykinin (960–1,680 pg/ml, measured according to (11, 12), with plasma from remission phase (279 pg/ml) tested in the same assay), all laboratory data including other coagulation factors were normal. During remission, levels of bradykinin, factor XII, C1inh and C1q were normalised (Table SI1).
DISCUSSION
To our knowledge, this is the first reported case of angioedema and urticarial vasculitis associated with autoimmune haemophilia. Months before the development of urticarial vasculitis and angioedema, the patient suffered from acquired haemophilia, which is an auto-immune disease caused by an inhibitory antibody to factor VIII. The disease occurs in about 1/1,000,000 individuals each year and ~20% of the cases are associated with other autoimmune diseases (10).
The case has many clinical similarities to hypocomplementaemic urticarial vasculitis with angioedema, but in contrast to this condition complement levels (C1-inhibitor and C1q) were increased during disease periods and no antibodies to C1q or C1-inhibitor were detected. Urticarial vasculitis is considered a type III hypersensitivity reaction in which immune complexes are deposited in the vessel walls leading to tissue damage. Angioedema is present in ~40% of patients with urticarial vasculitis (13, 14) of whom most have autoantibodies to C1q. Although our patient has some systemic lupus erythematosus (SLE) -like or SLE-associated features, such as a positive ANA and history of pericarditis and urticarial vasculitis, he did not fulfil the ACR criteria for SLE or any other autoimmune rheumatologic disease. Drug-induced disease could be ruled out since esomeprazole, the only drug apart from glucocorticoids the patient was taking when the symptoms started, had been discontinued several years before the patient had his relapse. Although measuring levels of bradykinin is known to be difficult due to the short half-live of the peptide, we believe that with optimal sampling and handling of the plasma we measured correct levels of bradykinin. Indeed high levels of bradykinin have previously been demonstrated in autoimmune diseases (11).
We hypothesise that the patient’s symptoms are related to an antibody-mediated malfunction of the kallikrein-kinin system, explaining the angioedema and elevated levels of bradykinin. Notably, autoantibodies that functionally interfere with enzymatic cascades have been described for thrombosis (15), glomerulonephritis (C3 nephritic factor), autoimmune haemophilia (for which the patient was treated) and acquired C1-inhibitor deficiency. It is noteworthy that the treatment of urticarial vasculitis with immunosuppressants dapsone, azathioprine and colchicine yielded complete recovery of the resistant angioedema, supporting the hypothesis that urticarial vasculitis and angioedema were signs of the same autoimmune disease. Off-label use of icatibant, a bradykinin B2 receptor antagonist, for the patient’s idiopathic angioedema (5) was considered, but was not available for treatment.This unusual case illustrates that malfunctions of the complement system is indeed not the only mechanism behind bradykinin-induced angioedema and urticarial vasculitis. The case highlights the need for more studies of the role of the contact/coagulation system in these diseases. Indeed, increasing evidence points at strong links between coagulation and inflammation (7–9), and current research is focused on elucidating the molecular mechanisms involved.
Acknowledgements
This study was supported by Queen Silvia’s Jubilee fond, the Royal Physiographic Society in Lund, Fanny Ekdahl’s Foundation and the Hedberg Foundation (to RK). We thank Prof. Diana Karpman (Department of Pediatrics, Lund University, for valuable discussions.

1http://www.medicaljournals.se/acta/content/?doi=10.2340/00015555-1909
References


