


1Department of Dermatology and Venereology, Aarhus University Hospital, Aarhus University, 2Department of Clinical Medicine, Aarhus University, Aarhus, Denmark, and 3Department of Dermatology, Radboud Institute for Molecular Life Sciences, Radboud UMC, Nijmegen, The Netherlands
The epidermal-derived “alarmins” high-mobility group box 1 (HMGB1) protein and interleukin-33 (IL-33) are upregulated in patients with atopic dermatitis. How-ever, their capacity as pro-inflammatory cytokines and their derived effects on skin barrier regulation are poorly elucidated. We investigated the impact of HMGB1 and IL-33 on gene transcription, protein expression and epidermal differentiation across 3 distinct keratinocyte in vitro models. Primary keratinocytes from healthy donors were used in submerged monolayer cultures, 3D human epidermis equivalents and 3D human skin equivalents. All keratinocyte models underwent 96-h stimulation with HMGB1 (100 μM) or IL-33 (100 ng/ml) using IL-4 (50 ng/ml) as positive control of regulation and vehicle as negative control. We found that HMGB1 and IL-33 downregulated transcription of several genes from members of the epidermal differentiation complex, including filaggrin. Furthermore, HMGB1 downregulated the expression of the encoded proteins in the upper epidermis. Finally, IL-33 and HMGB1 ultimately led to impaired epidermal growth and maturation. In conclusion, HMGB1 and IL-33 could play a significant role in the atopic dermatitis pathophysiology due to negative regulation of structural proteins, stratum corneum formation and epidermal growth.
Key words: filaggrin; involucrin; loricrin; IL-33; HMGB1; IL-4; skin; keratinocytes; cell culture; human skin equivalents; atopic dermatitis.
Accepted Oct 12, 2016; Epub ahead of print Oct 14, 2016
Acta Derm Venereol
Corr: Uffe Harboe Nygaard, Department of Dermatology and Venereology, Aarhus University Hospital, P.P. Oerumsgade 11, DK-8000 Aarhus, Denmark. E-mail: uffenygaard@clin.au.dk
The skin is a vital first line of defence between the environment and the human body. Studies indicate that defects in epidermal barrier function contribute to triggering and perpetuating skin inflammation in atopic dermatitis (AD) (1–5). Furthermore, disease activity and barrier function seem to be intimately related, and fluctuations in either can drive the disease (6). The role of skin barrier dysfunction as an important contributing factor in AD is further substantiated by loss-of-function mutations seen in the filaggrin gene which is a major risk factor for development of AD (3, 7, 8). Filaggrin is a key protein in epidermal homeostasis and barrier integrity; and through its processing to free amino acids, it facilitates retention of water in the stratum corneum (SC) (natural moisturising factor) (3, 9). In conjunction with filaggrin, other proteins encoded by genes in the epidermal differentiation complex, including loricrin and involucrin, are indispensable elements of the epidermal barrier (3, 10). Skin alterations in AD are characterised by increased transepidermal water loss (TEWL) and a defective terminal keratinocyte differentiation entailing reduced levels of filaggrin, involucrin and loricrin (11–13).
Loss-of-function mutations in the filaggrin gene are found in approximately one third of all AD patients and are considered responsible for the decreased skin barrier function, the increased TEWL and increased disease severity. Yet, all patients with AD display a reduced expression of filaggrin and also involucrin and loricrin. This phenomenon is explained in part by the impact of cytokines, e.g. interleukin (IL)-4, IL-13 and IL-25 that reduce the expression of barrier proteins (12–14).
The cytokine IL-33 is a member of the IL-1-family. IL-33 is constitutively expressed in healthy skin and is upregulated in skin of AD patients and in AD-like lesions in mice (15–18). Furthermore, we recently found elevated IL-33 serum levels in children with AD (19, 20). IL-33 is classified as an alarmin due to the passive release upon cellular stress like allergen stimulation, scratching or non-apoptotic cell death, though these mechanism are still to be fully elucidated (16, 21, 22). It displays Th2 immunomodulatory effects by inducing IL-5 and IL-13. It also displays a Th1 response via upregulation of IFN-γ (23). Keratinocytes produce IL-33 and also express its receptor complex consisting of ST2 and IL-1RAcP on their surface (16, 24). Moreover, IL-33 appears to exert receptor-independent transcriptional effects in the nucleus of epithelial cells (25), thereby operating as a dual-function immune mediator similarly to high-mobility group box 1 (HMGB1) (26). HMGB1 is localised in the nucleus in almost all cells and is rapidly mobilised to other sites within the cell, and into the extracellular space in response to a variety of stimuli (26, 27). Through RAGE and TLR4 signalling, HMGB1 mediates immune responses in non-infectious inflammation and shows upregulation in the skin and serum of AD patients (28, 29). HMGB1 sustains a long-term inflammatory state under stress similar to IL-33 by activating both keratinocytes and a range of immune-competent cells (e.g., macro-phages, monocytes and dendritic cells) which, in turn, form a positive feedback loop that causes the release of additional cytokines and chemokines (30–33).
Although we have in-depth understanding of the systemic actions of IL-33 and HMGB1, little is known about their direct effects on skin barrier integrity and homeostasis. In the present report, we investigate the impact of IL-33 and HMGB1 on primary human keratinocyte monolayer cultures and two different three-dimensional (3D) skin models. Our results demonstrate that HMGB1, and to a lesser extent IL-33, impair the gene transcription and protein expression of a range of pivotal barrier and epidermal differentiation proteins. Moreover, they reduce the epidermal growth and thereby hamper the formation of a normal, fully stratified epidermis. These results indicate that HMGB1 and IL-33 may impair skin barrier function and that they may be new targets in the intraepidermal pathophysiology of AD.
Primary human keratinocytes were collected and cultured as described previously (34). All keratinocyte monolayer cultures (KMCs) and stimulations were performed in duplicate. For the analysis of IL-33 and HMGB1 synthesis, primary keratinocytes (n = 4) were grown to about 60% confluency, stimulated with IFN-γ (5 ng/ml, #285-IF, R&D Systems), IL-25 (10 ng/ml or 100 ng/ml, #1258-IL, R&D Systems, Oxon, UK) or combinations of IFN-γ (5 ng/ml) and IL-25 (10 ng/ml or 100 ng/ml) for 24 h and collected in a lysis buffer for protein analysis.
For the evaluation of filaggrin, loricrin, involucrin, hornerin, SPINK5 and matriptase mRNA levels, cells (n = 5) cultured in growth medium supplemented with 1.3 mM calcium (Ca) to increase differentiation were stimulated for 4 days at 70-80% confluency with recombinant human IL-25 (10 or 100 ng/ml), IL-33 (2, 10 or 100 ng/ml, #3625-IL, R&D Systems),) or disulphide HMGB1 (100 μM, #HM-121, HMGBiotech, Milano, Italy). After 48 h, the medium supplemented with cytokine was refreshed. The cells were collected on day 4, washed in ice-cold phosphate-buffered saline (PBS) and total RNA was isolated using the SV Total RNA Isolation System (Promega, Madison, USA) as previously described (35).
Western blot analysis of IL-33 and HMGB1 synthesis in keratinocytes was performed as previously described (36). The following antibodies were used for evaluation of the protein expression: polyclonal goat anti-IL-33 (#AF3625; R&D Systems), polyclonal rabbit anti-HMGB1 (#ab18256, Abcam, Cambridge, UK) and horseradish peroxidase-conjugated mouse anti-human β-actin (Clone c4; Santa Cruz Biotechnology, Dallas, USA). Primary antibodies were visualised using horseradish peroxidase-conjugated polyclonal antibodies (p0449; DAKO, Glostrup, Denmark), (#7074, Cell Signalling Technology) and (p0447; DAKO). All samples were analysed in duplicate.
For RT-qPCR analysis, cDNA synthesis was performed using TaqMan reverse transcription reagents (Applied Biosystems, Waltham, USA). RPLP0 (Ribosomal protein, large, P0) was used as reference gene. PCR master mix and primers and probes for filaggrin, involucrin, loricrin, hornerin, matriptase, SPINK5 and RPLP0 were all commercially available (Taqman Master Mix #4369016, Taqman IDs: Hs00856927_g1, Hs00846307_s1, Hs01894962_s1, Hs02340614_m1, Hs01058386_m1, Hs00928575_m1 and Hs99999902_m1; all from Applied Biosystems). The probe was a FAM-labelled MGB probe with a non-fluorescent quencher. Each gene was run in triplicate on a StepOnePlus PCR system PCR platform (Applied Biosystems). The target gene expression level was normalised to the reference gene (RPLP0). The average of non-stimulated controls was set to 1, and the relative gene expression levels in comparison with the control were determined.
Primary keratinocytes were acquired from skin explants after abdominoplasty (n = 7). Isolation was done as described previously (37). Keratinocytes (1 × 105) were seeded onto polystyrene inserts (Thincert, Greiner bio-one, Kremsmünster, Austria) in proliferation medium (CnT-Prime, CellnTec, Bern, Switzerland). On day 2, the medium was replaced by a differentiation medium (3D barrier, CellnTec, Bern, Switzerland) complemented with DMEM (Gibco, Waltham, USA). Human epidermal equivalents (HEEs) were exposed to the air-liquid interface on day 3 to stimulate differentiation and stratification. The medium was refreshed every other day. HEEs were stimulated with IL-4 (50 ng/ml, #204-IL, R&D Systems), IL-33 (100 ng/ml) and disulphide HMGB1 (100 μM) on day 8. After 48 h, the medium supplemented with cytokine was changed. The HEEs were harvested for subsequent analysis on day 11. All cultures were antibiotics-free and handled with aseptic techniques in accordance with recommendations (19).
HEEs were fixated in a 4% buffered formalin solution and processed for routine histology. Paraffin sections of 6 µm were stained with haematoxylin and eosin to assess histology, while immunohistochemistry with indirect immunoperoxidase technique using avidin-biotin complex enhancement (Vectastain Laboratories, Burlingame, USA) was prepared to analyse epidermal differentiation. Antibodies against filaggrin (1:200; NCL-FLG, Leica Biosystems, Nussloch, Germany), loricrin (1:2000; BabCO, Covance, Princeton, USA), involucrin (1:20; Mon 150, custom made), keratin 10 (1:100; DE-K10, Eurodiagnostics, Apeldoorn, The Netherlands), keratin 14 (1:50; DE-K14, Eurodiagnostics), late cornified envelope 2 (1:1000; pan-anti-LCE2, custom made) and Ki67 (1:50; MIB-1; DAKO) were used and detected with 3-amino-9-ethylcarbazole (AEC). All immunohistochemical stainings were performed with a strict time protocol to avoid intra-analysis variance. Fixed software settings were used during histopathological imaging to ensure minimal slide-to-slide variability. An experienced technician blindly evaluated epidermal thickness and the number of ki67-positive cell nuclei.
RNA from duplicates was isolated in accordance with the manufacturer’s protocol (RLT buffer, QIAGEN, Valencia, USA). Likewise, cDNA synthesis and RT-qPCR analyses were performed according to the manufacturer’s recommendation (Biorad, Hercules, USA). Target gene expression was normalised to the expression of RPLP0, and relative expression levels were calculated by the ΔΔCt method (38). SYBR Green primer sequences are depicted in Table SI.
In-depth description of the development, culturing and handling human skin equivalent (HSE) is provided in detail in previous publications (39, 40). HSEs (n = 6) were stimulated on day 8 with IL-4 (50 ng/ml), IL-33 (100 ng/ml), disulphide HMGB1 (100 μM) or left unhampered. On day 10, the medium supplemented with cytokine was changed. The HSEs were harvested on day 11. Tissue fixation, processing for HE staining, RNA extraction and RT-qPCR analysis were performed similarly to the HEE procedures after dispase-facilitated isolation of epidermis (39).
The distribution of dependent data was assessed by histograms and Q-Q plots, which were supported by skewness and kurtosis tests for normality. Data that passed the normality test were analysed with unpaired t-test, paired t-test or one-way ANOVA, as appropriate. p-values < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001). Statistics were performed using STATA version 12.0 (StataCorp, College Station, USA) Graphs were prepared in GraphPad Prism version 6 (GraphPad software, La Jolla, USA).
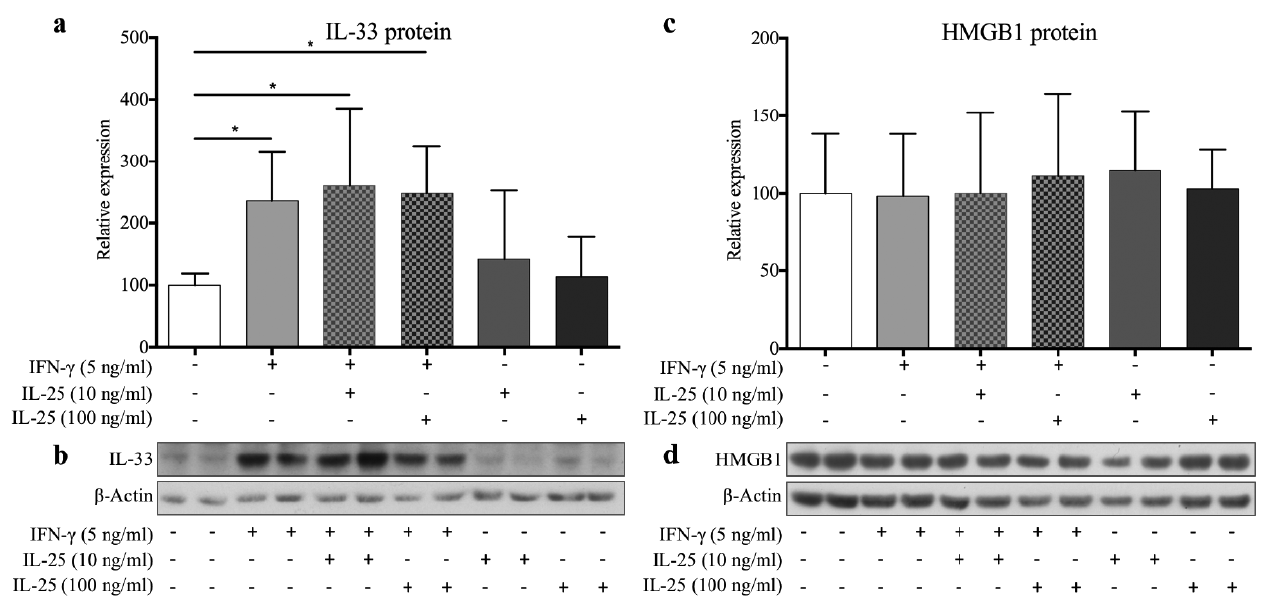
Our study revealed a significant relative increase in the IL-33 protein level when KCs were stimulated with IFN-γ compared with unstimulated controls. However, co-stimulation with IL-25 had no significant additive effect; nor did IL-25 alone affect IL-33 production (Fig. 1a, b). Studies of HMGB1 protein levels revealed only non-significant changes (Fig. 1, d).
Fig. 1. Interleukin (IL)-33 and high-mobility group box 1 (HMGB1) protein levels in keratinocytes stimulated with interferon (IFN)-γ and/or IL-25 (n = 4). (a) Densitometric band analysis of the Western blots showed significant IL-33 upregulation in all IFN-γ stimulations when compared with unstimulated control. (b) Representative IL-33 Western blots. (c) Quantified HMGB1 protein levels displayed no significant alterations. (d) Representative HMGB1 Western blots. Graphs show the relative (to unstimulated control) mean + standard deviation of measured protein levels. *p < 0.05 compared with control.
IL-33 was tested in a dose-response study with 2, 20 and 100 ng/ml. This test revealed a significant concentration-dependent downregulation of involucrin (p = 0.016), while the other targets were non-significant (data not shown). HMGB1 showed non-significant changes in a comparison time-response study (24 vs. 96 h) (data not shown).
KMCs were hence stimulated for 96 h with IL-25 (100 ng/ml), IL-33 (100 ng/ml) and HMGB1 (100 mM) which revealed a significant downregulation of filaggrin mRNA and involucrin mRNA and a non-significant regulation of loricrin, SPINK5 and matriptase mRNA compared with the relevant controls+Ca (Fig. 2). Controls without added Ca showed significantly lower mRNA levels of filaggrin, involucrin and SPINK5 than controls with added Ca.
Fig. 2. mRNA expression profiles in keratinocyte monolayer cultures measured by quantitative RT-PCR (n = 5). Filaggrin and involucrin mRNA levels were significantly downregulated following addition of interleukin (IL)-33 or high-mobility group box 1 (HMGB1). Graphs show the relative (to unstimulated control (–Ca)) mean + standard deviation (SD) of measured mRNA levels. *p < 0.05, **p < 0.01 compared with control+calcium (+Ca)).
Knowing that IL-33 and HMGB1 downregulated filaggrin and involucrin in KMCs, we wanted to interrogate the same hypothesis in an organotypic human epidermis model. We included the archetypical Th2 cytokine IL-4 as a positive control. Compared with the KMC studies, we excluded SPINK5 and matriptase due to an only modest sign of regulation from target cytokines, and included a wide range of other epidermal differentiation genes.
Our studies in HEEs revealed that stimulation with HMGB1 or IL-4 significantly downregulated the mRNA expression of filaggrin, involucrin, loricrin, LCE2A and KLK5. Furthermore, we found that IL-4 mediated a significant downregulation on the majority of additional targets (Table I). Most interestingly was the strong downregulation of residual structural protein genes, of kallikrein 5 and 7 and the highly significant upregulation of IL-33. Besides LCE2A mRNA, IL-33 produced no significant regulation of any of the examined differentiation genes, which is in contrast to the results obtained in the KMCs (Table I). Details on mRNA expression in individual genes are presented Fig. S1.
Table I. mRNA expression profiles in human epidermal equivalents (HEE) (n = 7) and human skin equivalents (HSE) (n = 6)
Concentrations and gene targets identical to those used in the HEE experiments were applied in the HSE study (Table I). HMGB1 produced downregulations paralleling those in the HEE model, in addition to hornerin, K10, and IL-18, and, interestingly, an increase in the IL-33 mRNA expression. Among other gene targets, IL-4 showed a consistent downregulation of all structural protein genes apart from loricrin. Similarly to the HEE analysis, a very significant upregulation of IL-33 mRNA was observed. IL-33 stimulation showed no significant regulation in the HSE model. Details on mRNA expression in individual genes are presented in Fig. S2.
As PCR analysis suggested a potential impact of the target cytokines on cell proliferation and differentiation, we consequently performed HE and Ki67 stainings to evaluate HEE growth parameters, and Ki67-positive nuclei.
The total epidermal thickness of the HEEs was significantly reduced in all stimulations (Fig. 3a). The thickness of the nucleated epidermal layers (NEL, total epidermal thickness without the SC) was reduced by IL-33 and HMGB1 stimulation, while the isolated SC thickness was significantly reduced by HMGB1 and IL-4 (Fig. 3b, c). The Ki67 analysis revealed a significant increase in the amount of Ki67-positive nuclei in all 3 stimulations (Fig. 4). Fig S3 displays representative Ki67 and HE stainings.
Fig. 3. Human epidermal equivalents (HEE) display significant morphological changes upon IL (interleukin)-33 (100 ng/ml), high mobility group box 1 protein (HMGB1) (100 μM) and IL-4 (50 ng/ml) stimulation (n = 7). (a) The total epidermal thickness of the HEEs was significantly reduced in all stimulations. (b) IL-33 and HMGB1 significantly reduced the thickness of nucleated epidermal layers (NEL) (total epidermal thickness without the stratum corneum (SC). (c) HMGB1 and IL-4 significantly reduced the thickness of SC. Graphs display the mean + standard deviation (SD) of measured thickness. *p < 0.05, ***p < 0.001 compared with control.
Fig. 4. Ki67-positive nuclei cell count in human epidermis equivalents is increased following interleukin (IL)-33, high mobility group box 1 protein (HMGB1) or IL-4 stimulation (n = 7). IL-33 (100 ng/ml), HMGB1 (100 μM) and IL-4 (50 ng/ml) all produced a significant increase in the quantity of Ki67-positive nuclei. The graph displays the mean + standard deviation (SD) of Ki67-positive nuclei cell count. **p < 0.01, ***p < 0.001 compared with control.
The protein expression of filaggrin in the upper epidermis was distinctly diminished by HMGB1 and IL-4 stimulation, while the supplement of IL-33 exhibited no apparent changes (Fig. 5). Involucrin staining was markedly decreased by IL-4, while HMGB1 produced a minor reduction and IL-33 no evident regulation (Fig. 5). HMGB1 and IL-4 stimulation reduced the protein expression of loricrin; conversely, IL-33 stimulation increased the expression (Fig. 5). Despite downregulation of LCE2A mRNA by all stimulations, immunohistochemical staining did not support a corresponding reduction of protein expression, which may, in part, be explained by the difficulty of LCE2 immunohistochemical interpretation (Fig S4). IL-4 stimulation resulted in a clear reduction of K10 expression in the HEE model, whereas other K10 and K14 stainings were inconclusive compared with unstimulated controls (Fig. S4).
Fig. 5. The protein expression of filaggrin, involucrin and loricrin in the upper epidermis of human epidermis equivalents (HEE) is significantly regulated by interleukin (IL)-33, high mobility group box 1 protein (HMGB1) or IL-4 stimulation (n = 7). Representative immunohistochemical stainings of filaggrin, involucrin and loricrin upon IL-33 (100 ng/ml), HMGB1 (100 μM) or IL-4 (50 ng/ml) stimulation. The protein expression of filaggrin in the upper epidermis was distinctly diminished by HMGB1 and IL-4 stimulation. Involucrin staining was markedly decreased by IL-4, while HMGB1 produced a minor reduction. HMGB1 and IL-4 stimulation reduced the protein expression of loricrin; conversely, IL-33 stimulation increased the expression. Scale bar = 100 μm.
HSE growth and differentiation were validated from HE staining presented in the online supplement (Fig. S5). Immunohistochemical stainings of HSEs were of little value because they displayed lesser upper epidermal differentiation than HEEs which made the staining weaker and challenging to interpret (stainings not shown).
AD is characterised by a dysregulation of several cytokines, and it has been suggested that these cytokines participate in disease pathophysiology (41, 42). Two of the effector cytokines possibly implicated in AD are the ”alarmins” IL-33 and HMGB1. Other well-described cytokines, including IL-4, IL-13 and IL-25 (12-14, 16, 28), may also be implicated. Here, we demonstrate for the first time that HMGB1 and IL-33 downregulate transcription of genes from members of the epidermal differentiation complex. Furthermore, HMGB1 downregulates the expression of several of the encoded proteins in the upper epidermis. Finally, we show that IL-33 and HMGB1 stimulation ultimately leads to impaired epidermal growth and maturation. While HMGB1 shows uniform effects across 3 keratinocyte models, the impact of IL-33 is less pronounced.
The inflammatory response mediated by HMGB1 is well characterised across a broad palette of cells and tissues (26). However, the role of HMGB1 in skin barrier homeostasis and AD is poorly elucidated. HMGB1 expression is chronically augmented during various types of skin inflammation, including AD (28, 29, 43–46). Our results give us reason to believe that constant HMGB1 stimulation of keratinocytes within AD lesions can impair natural barrier formation. Keeping in mind that the HMGB1 concentrations used in our study could enhance any effects compared with the pathophysiological situation, we show that HMGB1 heavily impacts the gene products of pivotal skin barrier differentiation genes, e.g. filaggrin, involucrin and loricrin. Consequently, HMGB1 has deleterious effects on protein expression, epidermal growth and SC formation. This is in line with the effects of HMGB1 in lung and intestinal endothelia cells where HMGB1 gives rise to cytoskeletal rearrangement and barrier disruption (47, 48). We show an upregulation of Ki67 in the basal cell layer in HMGB1-stimulated cells that does not, as would be expected, result in increased epidermal growth and barrier maturation; in fact, we observe the opposite. These results are in concert with previous in vitro studies of HMGB1 as increased Ki67 expression and cell proliferation of adult and embryonic mesoangioblasts disrupt the barrier function of endothelial monolayers (49). Whether the impact of HMGB1 in the AD in vivo setting might mimic that seen in psoriasis need further exploration (45).
IL-4 and HMGB1 experiments revealed a recurrent downregulation of kallikrein 5 mRNA. This regulation may hint at a meaningful counter response to the deterioration of skin barrier formation as it is recognised that increased levels and activity of kallikrein 5 are important to the development and maintenance of AD. However, these possible dual functions need further study (50, 51). Additionally, in the 3D models HMGB1, in union with IL-4 showed a highly significant upregulation of the IL-33 mRNA product that parallels results reported in studies of other epithelial cells (52). This could indicate the existence of a damaging feedback loop enhancing any IL-33 properties, e.g systemic Th2 skewing TSLP and IL-13 upregulation in the skin, augmenting ongoing immunomodulation, diminishing barrier function and possibly reducing the effect of topical glucocorticosteroid treatment in AD patients (23, 53–56).
IL-33 is reported to be elevated in both the blood and epidermis of AD patients (16, 17, 20). Our study verified the IL-33 production from keratinocytes under IFN-γ exposure. It also revealed an IL-33 upregulation after HMGB1 or IL-4 stimulation, and it disclosed a downregulation of filaggrin and involucrin mRNA in the KMCs upon IL-33 stimulation, although no regulation was observed in either of the 3D models. These findings are partly in line with those of a previous study investigating similar causality (57). Seltmann et al. showed significant downregulation of filaggrin mRNA in lesional AD keratinocytes upon IL-33 stimulation. Still, they did not reproduce these results in normal human KMCs, AD skin biopsies or in a HEE model, data mirrored in our study. Lastly, and in contrast to our results, they reported a reduction in filaggrin protein expression by IL-33 evaluated through IHC staining of HEEs. The fact that we disclosed no effect of IL-33 on isolated SC thickness support our mRNA data, as neither of the eight gene targets representing the bulk mass of protein in the SC, was significantly regulated in either the HEE- or HSE mRNA profile. Together these findings give little reason to believe that IL-33 heavily impacts structural skin barrier protein expression and translation. However, the question remains unanswered whether IL-33 does, in fact, negatively impact production and maturation of filaggrin, involucrin and other structural barrier proteins. Our studies of HEE thickness support the evidence of an intraepidermal role of IL-33. We show a clear reduction of epidermal growth upon IL-33 stimulation despite an increased Ki67-positive cell count. This is possibly due to mechanisms mimicking those of HMGB1 where significant upregulation of Ki67 and cell proliferation do not translate into increased growth, but instead hamper epidermal development (49). This causality is further supported by the increased production of IL-33 from lung epithelial cells causing a reduced alveolar diffusion capacity and aggravation of chronic obstructive lung disease (58).
Several papers describe the downregulatory effects of IL-4/IL-13 concerning keratinocyte proliferation, differentiation and filaggrin expression (12, 13, 57, 59–61). Our present report offers a consolidation of the isolated impact of IL-4 on filaggrin, involucrin and loricrin gene and protein expression. Additionally, it is also the first study exploring the effect of IL-4 in HEEs supported by two additional models; and it furthermore shows impaired HEE growth and SC formation, thereby accentuating the prospect of the ongoing studies of AD patients treated with dupilumab, a fully human monoclonal antibody that blocks the IL-4 receptor subunit IL-4Rα, hence modulates signalling of both the IL-4 and IL-13 pathway (62, 63).
In the present study, we provide a triple in vitro study of the impact of HMGB1, IL-33 and IL-4 on the most pivotal epidermal differentiation genes, the protein expression in a HEE model and the evaluation of HEE growth and SC formation. Furthermore, we report a consistent and convincing reduction of hallmark skin barrier gene and protein products upon stimulation with HMGB1 and IL-4. We speculate that HMGB1 has primary regulatory effects on keratinocyte homeostasis and could be a major target for future research into barrier dysfunction in AD. IL-33 has only discrete gene and protein regulatory effects; however, its capacity to decelerate normal epidermal growth might turn out to be an important part of the negative feedback loop of the intraepidermal pathophysiology of AD.
This study was supported by Kgl. Hofbuntmager Aage Bangs Fond, OTICON Fonden, Carl og Ellen Hertz’ Legat, Dagmar Marshall Fond, Hørslev Fonden.
The authors declare no conflict of interest.


 Click to show fullsize
Click to show fullsize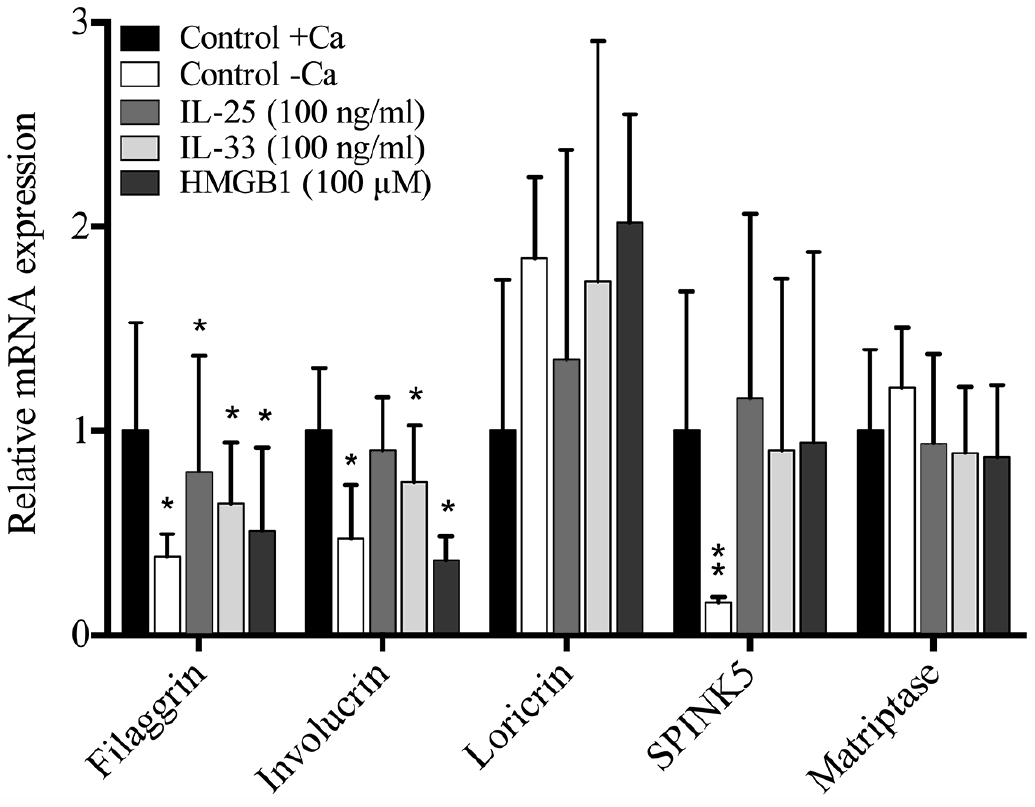 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize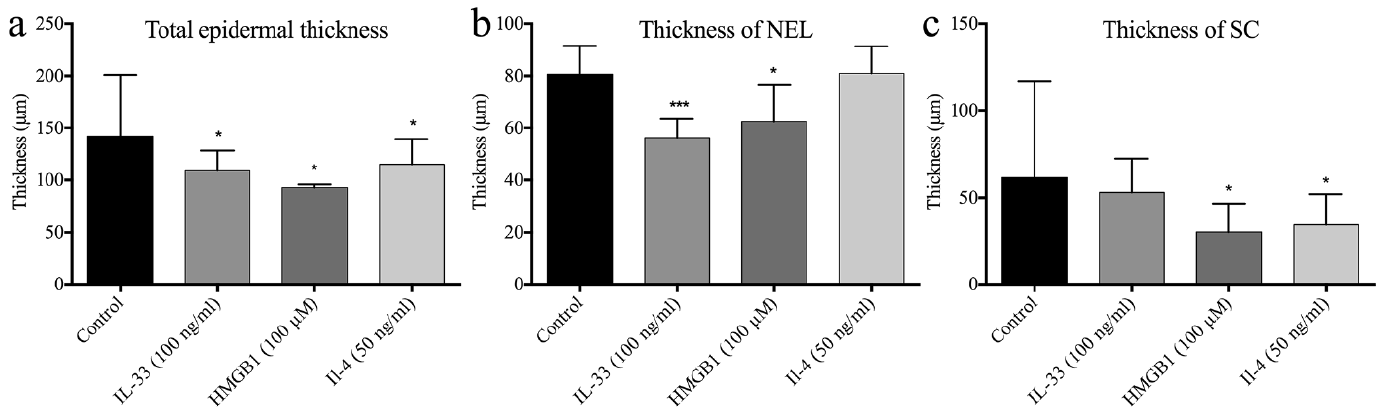 Click to show fullsize
Click to show fullsize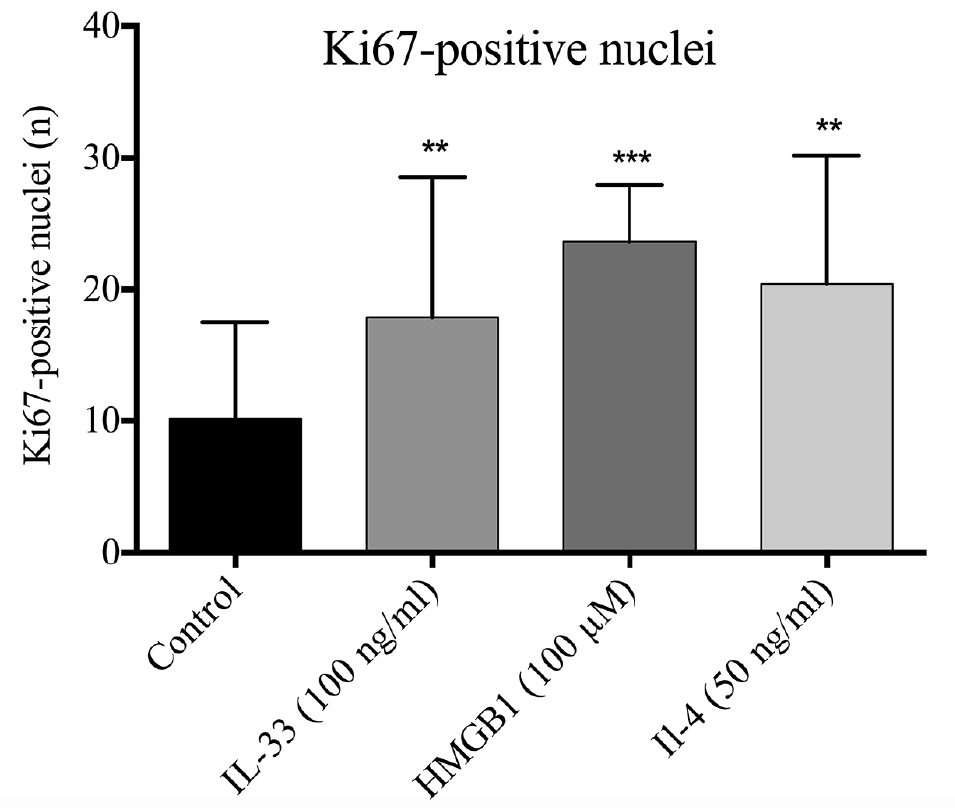 Click to show fullsize
Click to show fullsize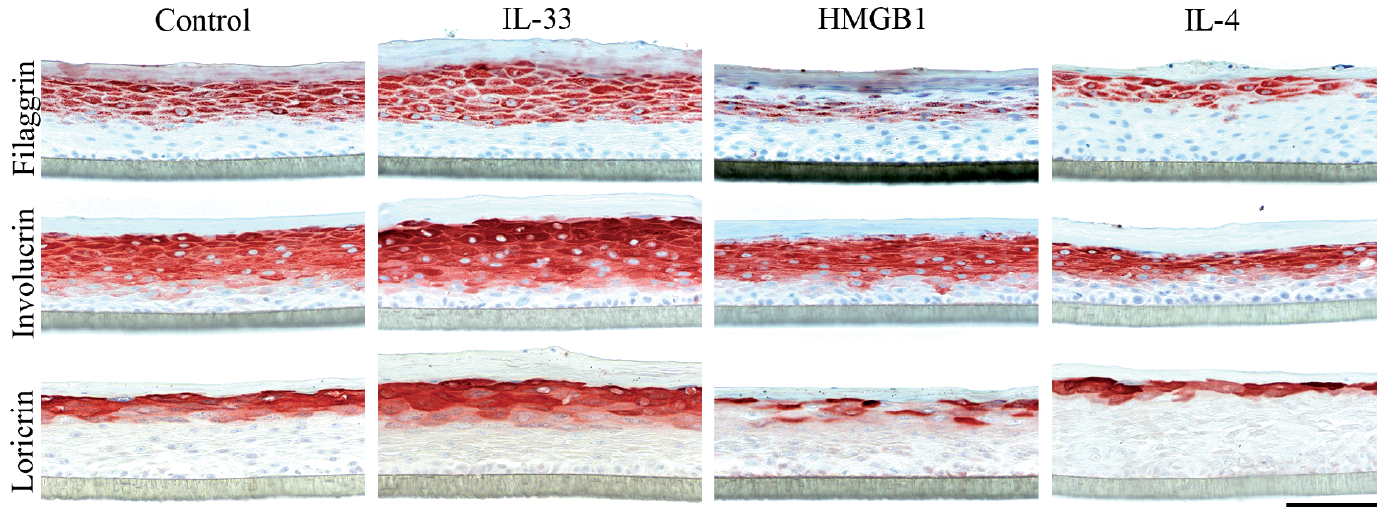 Click to show fullsize
Click to show fullsize