


Departments of 1Dermatology, 4Plastic Surgery, 6Pathology and 7Pediatric Hemato-Oncology and Bone Marrow Transplantation, Pediatric Division, Hadassah-Hebrew University Medical Center, Jerusalem, Departments of 2Dermatology and 3Pediatric Hemato-Oncology and Bone Marrow Transplantation, The Edmond and Lily Safra Children Hospital, Sheba Medical Center, Tel-Hashomer and Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv and 5Hadassah-Hebrew University Faculty of Medicine, Jerusalem, Israel
#These authors contributed equally to this study.
Chronic graft versus host disease (cGVHD) is a complication of allogeneic haematopoietic stem cell transplantation (HSCT). The aim of this study was to clinically characterize childhood cutaneous cGVHD. A retrospective study of children treated with HSCT at 2 tertiary medical centres in Israel between 2011 and 2014 was performed. A total of 112 children were included. Cutaneous cGVHD developed in 18% of subjects. Risk factors were older age, HSCT from peripheral blood and acute lymphoblastic leukaemia. The eruption was lichenoid in 90% of subjects, of whom one-third progressed to sclerosis. Topical treatments were usually sufficient in localized disease. Widespread eruption necessitated phototherapy, extracorporeal photopheresis and/or systemic immunosuppressants. Patients presenting with palmoplantar keratoderma, developed sclerosis. To the best of our knowledge, this is the first study describing childhood cutaneous cGVHD. Lichenoid eruption is the most common cutaneous pattern of cGVHD in children. Sclerotic changes may be associated with prior keratoderma. cGVHD poses a therapeutic challenge and better treatments should be sought.
Key words: allogeneic haematopoietic cell transplantation; graft versus host disease; chronic cutaneous graft versus host disease; children; lichenoid; sclerotic.
Accepted Oct 19, 2017; Epub ahead of print Oct 23, 2017
Acta Derm Venereol 2018; 98: XX–XX.
Corr: Vered Molho-Pessach, Department of Dermatology, Hadassah-Hebrew University Medical Center, Kiryat Hadassah, POB 12000, Jerusalem, 9112001, Israel. E-mail: Rverem@hadassah.org.il
Allogeneic haematopoietic stem cell transplantation (HSCT) is a life-saving treatment for haemato-oncological malignancies, hereditary disorders and primary immuno-deficiencies. The use of allogeneic HSCT in children has been expanding in recent years. Conditioning by chemotherapy with or without irradiation is given to destroy the immune system. This is followed by transplantation of haematopoietic stem cells harvested from the donor’s peripheral blood stem cells (PBSC), bone marrow or using umbilical cord blood (1). The donor can be a family member or an unrelated donor, and the degree of matching is classified according to the human leukocyte antigen (HLA) system (2).
Excluding infections, graft versus host disease (GVHD) is the most common complication following allogeneic HSCT, and is a major cause of morbidity and mortality. GVHD can affect any body organ, but the skin is most commonly involved (1). GVHD occurs when the donor’s immune cells recognize the recipient’s tissues as foreign due to an interaction between the recipient’s antigen-presenting cells and the donor’s mature T cells, leading to immune dysregulation (3). Nevertheless, GVHD is also considered beneficial in haematological malignancies, due to an immunological effect of the donor’s immune system against the tumour cells (4). GVHD is divided into acute and chronic reactions, traditionally distinguished by a cut-off of 100 days following HSCT. Nowadays, the distinction is defined by specific clinical features and by different pathophysiology (5). Acute GVHD (aGVHD) is caused by reaction of the host’s dendritic cells to the graft, which results in activation of donor-derived T cells, activation of type 1 helper T (Th) cells and tissue damage (6). Clinically there is a triad of skin eruption, hyperbilirubinaemia and diarrhoea (7). Chronic GVHD (cGVHD) involves allogeneic and autoimmune-like reactions. The conditioning regimen and/or aGVHD damages the thymus and impairs self-tolerance. CD4 and CD8 T cells, regulatory T cells and B cells participate in the production of autoantibodies. Activation of Th1, Th2 and Th17 cells results in tissue inflammation, leading eventually to fibrosis (8–10). Cutaneous involvement is observed in 75% of cases, followed by hepatic, oral, ophthalmic or lung involvement (11). Recognized risk factors are: older age of the patient or donor, former aGVHD, unrelated donor with low HLA matching, use of progenitor haematopoietic cells derived from PBSC, a diagnosis of chronic myeloid leukaemia, and infusion of donor white blood cells (6, 12). Cutaneous features of cGVHD are pleomorphic. Typically non-sclerotic and sclerotic forms are distinguished (1). The non-sclerotic form is usually lichenoid (simulating lichen planus), but other less common morphologies have been described, such as keratosis pilaris-like, ichthyotic-like, poikilodermatous, papulosquamous, acral erythema and eczema/atopic dermatitis-like (1, 13, 14). The sclerotic form may result in joint contractures. Lichenoid eruptions tend to appear earlier and may evolve into sclerotic lesions, although these may appear without a preceding lichenoid phase (1, 13). The diagnosis of cGVHD is based on clinical features. According to the National Institutes of Health (NIH) Consensus Development Project, the following skin manifestations are diagnostic of chronic GVHD: poikiloderma, lichen planus-like eruptions, deep sclerotic features, morphea-like superficial features, and lichen sclerosus-like lesions (11).
Treatment of cGVHD consists of topical cortico-steroids and calcineurin inhibitors and, at times, photo-therapy (as narrow-band ultraviolet B (nbUVB) or psoralen plus UVA or UVA1 in sclerotic changes) in mild cutaneous disease. Sclerotic or widespread disease is more resistant to treatment and may require the use of extracorporeal photopheresis (ECP), systemic cortico-steroids, immunosuppressants, or biological treatments (15, 16).
Despite the increased use of allogeneic HSCT in the paediatric population in recent years, there are sparse data regarding the prevalence, risk factors, clinical features, course, and response to treatment of cutaneous cGVHD in children (17).
The aim of this study is to characterize cutaneous cGVHD following allogeneic HSCT in the paediatric population.
This is a retrospective study of children who underwent HSCT at the paediatric haemato-oncology and bone marrow transplantation departments at 2 tertiary medical centres in Israel: Hadassah-Hebrew University Medical Center and Sheba Medical Center. These 2 medical centres differ mainly by the origin of the haematopoietic stem cells; PBSCs are used more frequently at Sheba Medical Center, while bone marrow is usually used at Hadassah-Hebrew University Medical Center. Patients included were 0–18-year-olds, who underwent allogeneic HSCT between 2011 and 2014.
Information collected from the medical records included sex, age at transplantation, diagnosis of primary disease requiring transplantation, type of transplantation and donor, complications of transplantation and mortality, dermatological manifestations and therapy. Cutaneous GVHD was divided into acute and chronic, according to clinical features. The diagnosis of cutaneous cGVHD and additional cutaneous findings were performed and recorded by a dermatologist. Reports of skin biopsies, if performed, were reviewed.
Collection and analysis of data started in September 2015 and ended by June 2016. The follow-up period was from diagnosis of cGVHD to the end of the study, or the death of patients. The rate of missing data was low and limited to one patient with cGVHD (patient 15). All data were summarized and displayed as a number (percentage) of patients in each group for categorical variables and as a mean ± standard deviation (SD) for continuous variables. Continuous variables were compared using the independent samples t-test or analysis of variance (ANOVA) (Scheffee was used for post-hoc multiple comparison), while categorical variables were compared using a Fisher’s exact test. All analyses were performed with SPSS 21.0 software (SPSS Inc., Chicago, IL, USA).
This study was approved by the Hadassah-Hebrew University Medical Center and the Sheba Medical Center institutional review boards.
A total of 112 patients were included in the study. Demographic and clinical characteristics are described in Table I. There were 73 males (65%) and 39 females (35%), with a mean age at transplantation of 6.05 years. Most of the patients (61%) were of Arab ethnicity. Acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL) were the most common diagnoses requiring HSCT, comprising 41% of the study population. The second most common diagnosis was primary immunodeficiency (19%). Most of the transplantations were performed from bone marrow as a graft source (80%) with an HLA-matched sibling donor (MSD) in 52% of the study population.
Table I. Characteristics of the study population (n = 112)
Cutaneous GVHD was found in 29% of the study population, composed of 19% of aGVHD and 18% of cGVHD. Of those patients with cutaneous cGVHD (n = 20), 9 patients progressed to cGVHD after a phase of aGVHD, while 11 patients developed cutaneous cGVHD de novo (Table II). The mean ± standard deviation (SD) follow-up period was 27.4 ± 13.5 months. In comparison between all patients with cutaneous GVHD (acute and/or chronic) and patients without cutaneous GVHD, a significant difference was found regarding age at transplantation, type of transplantation and donor (Table III). The mean age at transplantation was older in the cutaneous GVHD group (p = 0.023), in addition to a higher probability of peripheral blood HSCT (odds ratio (OR) 4.4, 95% confidence interval (95% CI) 1.41–16.31, p = 0.007) and matched unrelated donor (MUD) (OR 2.65, 95% CI 1.02–6.79, p = 0.041) in comparison with the non-cutaneous GVHD group.
Table II. Proportions of graft versus host disease (GVHD) in the study population (n = 112)
Table III. Comparison between patients with acute and/or chronic cutaneous graft versus host disease (GVHD) and without cutaneous GVHD
Twenty patients developed cutaneous cGVHD (18% of all transplanted children), 13 males and 7 females, with a mean age at transplantation of 9.5 years. The mean ± SD time interval from transplantation to the appearance of dermatological signs and symptoms was 4.9 ± 3.95 months, with a mean ± SD follow-up period of 27.9 ± 13.0 months. As shown in Table IV, when comparing patients with cutaneous cGVHD and patients without cutaneous GVHD, a significantly older age at transplantation was demonstrated (t(98) 3.35, p = 0.012). HSCT with bone marrow as a graft source was associated with a lower risk of cutaneous cGVHD (OR 0.18, 95% CI 0.05–0.59, p = 0.02), while PBSC HSCT was associated with higher risk (OR 10.43, 95% CI 3.12–34.36, p < 0.01). No significant difference was found comparing the type of donor. The diagnosis associated with greater probability for cutaneous cGVHD was ALL (OR 5.15, 95% CI 1.55–16.83, p = 0.003).
Table IV. Comparison between patients with cutaneous chronic graft versus host disease (cGVHD) and without cutaneous GVHD
Table SI describes the clinical characteristics of cutaneous cGVHD. The most common eruption was lichenoid (Table SI, Fig. 1), observed in 18 patients (90%). In 2 patients, the lichenoid rash was photo-distributed. Six patients with lichenoid eruptions progressed to develop sclerotic changes (Fig. 2) in a mean ± SD time interval of 10 ± 2.12 months following the lichenoid eruption and 19.6 ± 12.17 months following the HSCT. We did not observe the appearance of sclerotic changes without a preceding lichenoid phase. One patient presented with an eczematous eruption and one patient had an ichthyosiform eruption. Nine patients had localized rash, involving less than 50% of body surface area (BSA), 10 patients had widespread rash, involving more than 50% of BSA (data was missing for one patient). All patients with sclerotic changes had a prior widespread lichenoid rash. None of the patients with localized rash progressed to develop sclerotic changes, and in 4 patients (20%) with sclerotic cGVHD there were joint contractures.
Fig. 1. Lichenoid chronic graft versus host disease. (a) The trunk and (b) the arm in patient number 10.
Fig. 2. Sclerotic chronic graft versus host disease. (a) The abdomen and (b) the back in patient number 4.
In 6 patients (patients 1, 5, 8, 11, 14 and 20) a skin biopsy was performed, demonstrating findings that were compatible with cGVHD, such as vacuolar interface dermatitis and dermal melanophages. Additional cutaneous findings included palmoplantar keratoderma (Fig. 3) in 4 patients (20%), including one patient who demonstrated extremely hyperkeratotic lesions on the palms (Fig. 3c). The possibility of squamous cell carcinoma was suspected and therefore a skin biopsy was performed. This exhibited compact hyperkeratosis without atypia in all epidermal layers, suprabasal separation of the epidermis, focal basal vacuolar degeneration, as well as dermal fibrosis and mild perivascular inflammatory infiltrate. These changes are compatible with cGVHD (Fig. 3d) and rule out non-melanoma skin cancer. All patients with keratoderma eventually progressed to develop sclerotic cGVHD. One patient with sclerotic cGVHD (patient 14) developed erosions in sclerotic areas. Scarring alopecia was observed in 5 patients (Fig. 4) and nail changes (periungual violaceous plaques, nail dystrophy, pterygium) in 15% (Fig. 5). Further cutaneous findings unrelated directly to cutaneous cGVHD were plantar eccrine poromas, in one patient. Non-melanoma skin cancers were not observed in this group of patients.
Fig. 3. Keratoderma. (a) The palms and (b) the feet in patient number 10. (c) Clinical and (d) histopathological findings of a keratotic lesion of the palm in patient number 5. (d) Haematoxylin & eosin ×10; showing compact hyperkeratosis with focal vacuolar degeneration of the basal layer, serum and blood in the hyperkeratotic epidermis, as well as suprabasal separation of the epidermis and dermal fibrosis with few mononuclear cells.
Fig. 4. Alopecia. (a) Scarring alopecia and (b) its progression after 2 months of follow-up in patient number 10.
Fig. 5. Nail changes. (a) Periungual violaceous plaques, keratoderma, (b) pterygium and dystrophic changes after 19 months in patient number 10.
The treatment given for cGVHD is shown in Table SI1. Most patients were treated topically with emollients, corticosteroids and topical calcineurin inhibitors. All 9 patients with localized disease responded to topical treat-ment; however, 5 of them necessitated further therapy with ECP and/or systemic immunosuppressives due to involvement of other organs. As for widespread disease, 9 patients were treated topically, but eventually required additional treatments, including phototherapy, ECP and systemic agents. Phototherapy (UVA1 and nbUVB) was used in 4 patients (20%) with widespread cGVHD. Three patients, treated with nbUVB, received this treatment for a widespread lichenoid eruption, with transient improvement, but eventually progressed to develop sclerotic changes. One patient treated with UVA1 for sclerotic cGVHD experienced clinical improvement in her cutaneous symptoms, documented by ultrasonographic imaging (patient 3). Ten patients (50%) received ECP, but in only 4 of them it was given to treat widespread cutaneous cGVHD (patients 4, 7, 8, 14) without clinical improvement. Fifteen patients (75%) needed oral systemic treatment, mainly to treat involvement of organs other than the skin. However, only 5 of them (patients 4, 7, 8, 10, 14), required systemic therapies for their skin condition, mainly for widespread sclerotic changes. Prednisone was given to 15 patients. Other immunosuppressants were azathioprine, cyclosporine, imatinib, metho-trexate, tacrolimus, intravenous lidocaine, etanercept, bortezomib, mycophenolic acid, and mesenchymal stem cells. Regarding the efficacy of the various treatments, we related only to the 5 patients who received it for their cutaneous disease. Of these, only one patient (patient 8), improved on oral systemic therapy, which included mycophenolic acid and tacrolimus, given in combination with ECP. The remaining 4 patients did not respond to multiple systemic immunosuppressants and progressed to severe sclerotic disease with functional impairment.
Nail changes and scarring alopecia were improved in patients with localized disease, while in patients with widespread disease that required systemic therapies these changes progressed over time (Figs 4, 5).
The overall survival was 85%, with 3 (14%) cases of mortality due to septic shock or relapse of the primary disease. The cutaneous eruptions were not directly related to the cause of death.
This is the first study to describe cutaneous cGVHD in children. Among the 112 children undergoing allogeneic HSCT in 2 tertiary medical centres in Israel, 32 patients (29%) developed either aGVHD or cGVHD, affecting the skin.
Cutaneous cGVHD was observed in 18% of all transplanted children. The prevalence rates of cutaneous cGVHD in the adult population is unclear due to varied HSCT protocols, but is presumed to be higher (75% among those who develop cGVHD) (18). Risk factors associated with cutaneous cGVHD in our study were: older age, use of PBSC as a graft source as opposed to bone marrow cells, and ALL. Prior aGVHD and type of donor did not reach statistical significance. It is likely that ALL was a risk factor due to the fast tapering down of post-transplant immune suppression in these patients, done to produce the desired graft versus leukaemia effect. An additional possible factor is the use of total-body irradiation in the conditioning regimen in ALL patients, which results in higher toxicity and end organ damage.
The most common cutaneous pattern of cGVHD was lichenoid eruption, observed in 90% of patients. It was widespread in approximately half of these. Data regarding the prevalence of various cGVHD morphologies in adults is lacking, since differentiating clinical features are often not reported (1). We observed a tendency for localization of limited lichenoid eruptions to the face, scalp and upper trunk. It may be related to photo-distribution, which was noticed in 2 patients.
Thirty percent of patients with cutaneous cGVHD (5% of total transplanted children), developed sclerosis. A recent study found a cumulative risk of sclerosis of 20% at 3 years post-transplantation in 977 adults and children, data regarding rate of sclerosis among children was not given (19). Sclerotic changes in our study were observed only in patients with prior widespread lichenoid eruptions and appeared in approximately half of the patients with widespread lichenoid eruptions. Other morphological features observed were eczematous and ichthyosiform eruptions, palmoplantar keratoderma, scarring alopecia and various nail abnormalities. Keratoderma was observed during the lichenoid stage only in patients who eventually developed sclerosis. Therefore, keratoderma may be a sign of future cutaneous cGVHD progression. We did not detect any case of non-melanoma skin cancer or melanoma, as opposed to an increased risk of cutaneous malignancy recently reported in adults undergoing HSCT (20). This is probably related to the young age of our patients, lack of chronic sun exposure at this age and short follow-up period. We did, however, observe the appearance of recurrent plantar eccrine poromas in one patient. Eccrine poromatosis has been rarely described in patients with cGVHD and has been associated with human papillomavirus infection (21).
As for treatment, assessing the efficacy of various treat-ment modalities is problematic due to the small sample size, non-uniform treatment approaches between the 2 centres, as well as within each centre, and the fact that treatment was also guided by extra-cutaneous involvement. Nevertheless, it seems that patients with localized cutaneous cGVHD responded well to topical treatment. Patients with widespread involvement necessitated phototherapy, ECP and/or systemic treatments. nbUVB resulted in merely temporary improvement, while UVA1 was found to be effective in one patient with sclerotic cGVHD. ECP was effective in one patient in combination with systemic immunosuppression. Systemic agents, given for cutaneous disease in 5 patients, were effective in only one patient, while the remaining 4 patients did not respond to multiple treatments and eventually developed widespread sclerotic changes resulting in impaired joint mobility.
Our study has several limitations. First, this is a retrospective study, which may result in incomplete documentation. Secondly, conditioning regimens, treatment for GVHD prophylaxis, as well as for GVHD, were diverse. This may have affected the rate of cutaneous cGVHD and its clinical features.
Further prospective large-scale studies are necessary to define a treatment algorithm for children with cutaneous cGVHD. Cutaneous cGVHD, mainly of the sclerotic type, still poses a therapeutic challenge, and better treatments should be sought for this debilitating condition.
This research was approved by Hadassah-Hebrew University Medical Center and Sheba Medical Center IRB institutional board committees.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize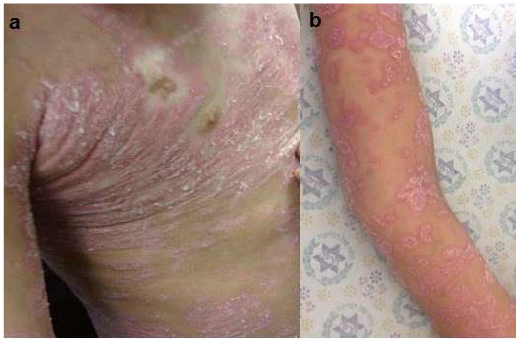 Click to show fullsize
Click to show fullsize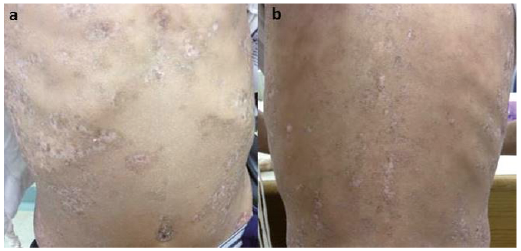 Click to show fullsize
Click to show fullsize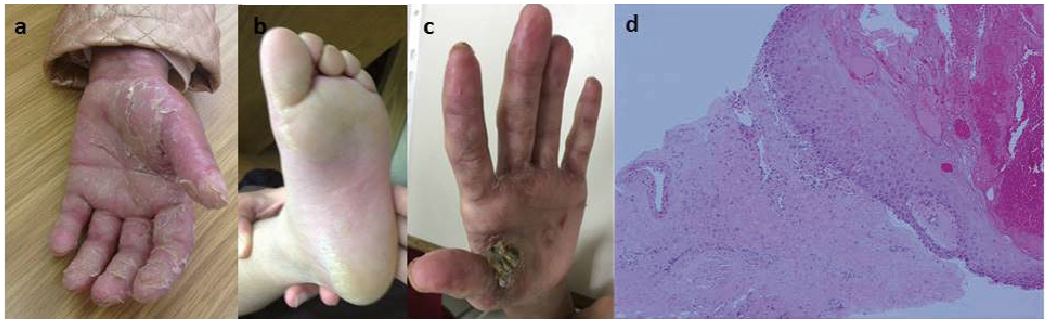 Click to show fullsize
Click to show fullsize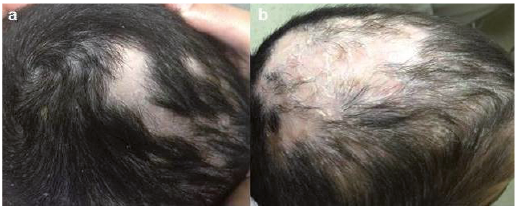 Click to show fullsize
Click to show fullsize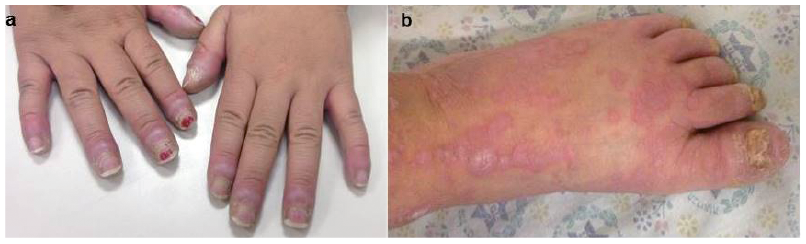 Click to show fullsize
Click to show fullsize