


1Department of Dermatology, 2Institute for Human Genetics, Medical Center, University of Freiburg, and 3Berta-Ottenstein-Program, Faculty of Medicine, University of Freiburg, Freiburg, Germany
#These authors contributed equally to this paper.
The precise classification of epidermolysis bullosa (EB) into 4 main types and more than 30 subtypes is based on the level of skin cleavage, as well as clinical and molecular features, and is crucial for early prognostication, case management, genetic counselling and prenatal or pre-implantation diagnosis. We report here the molecular pathology of 40 consecutive cases of suspected EB, which were investigated by immunofluorescence mapping (IFM) and/or by a targeted next-generation sequencing (NGS) multi-gene panel. IFM correctly established the EB subtype in 76% of cases, while the molecular pathology was completely elucidated in 90% of cases by the targeted NGS multi-gene panel. Thirteen previously unreported mutations in EB genes were identified. In cases with unclear clinical and IFM findings, mutations were found by NGS in previously unexpected genes. IFM was useful in delivering fast results in newborns, and in indicating the consequences of the variants of uncertain significance on protein level. This study underscores the efficacy of the strategy of combining targeted NGS with IFM in resolving unusual EB phenotypes. It also suggests that, despite technological advances, careful clinical evaluation and deep phenotyping remains a crucial factor that dictates successful diagnosis of EB.
Key words: epidermolysis bullosa; adhesion; blistering; skin fragility; mutation; keratin; collagen.
Accepted Dec 14, 2017; Epub ahead of print Dec 15, 2017
Acta Derm Venereol 2018; 98: xx–xx.
Corr: Cristina Has, Department of Dermatology, Medical Center, University of Freiburg, Faculty of Medicine, University of Freiburg, Hauptstrasse 7, DE-79104 Freiburg, Germany. E-mail: cristina.has@uniklinik-freiburg.de
Epidermolysis bullosa (EB) is a clinically and genetically heterogeneous group of disorders characterized by skin fragility and mechanically induced skin blistering. The genetic basis of EB is well-established (1), yet new genes and mutational mechanisms still emerge. Classification of EB into 4 main types (simplex, junctional, dystrophic and Kindler syndrome) and more than 30 subtypes is based on immunofluorescence mapping (IFM) and mutation analysis (2). Precise classification is crucial for early prognostication, case management, genetic counselling and prenatal or preimplantation diagnosis (2). Classical types of EB, such as severe generalized dystrophic or junctional EB, can be recognized based on clinical criteria as soon as the clinical picture is fully manifest (3). In newborns, infants and in individuals with moderate or mild skin fragility, clinical assessment usually does not allow precise classification. Such cases require molecular and genetic diagnosis.
We report here the molecular pathology of 40 consecutive suspected cases of EB, which were investigated by IFM and/or by a targeted next-generation sequencing (NGS) multi-gene panel since July 2016, when NGS was approved as a diagnostic method in Germany.
IFM was performed with a panel of 17 antibodies to components of the dermal–epidermal junction, as described previously (4). The multi-gene panel included 49 genes: 19 genes in which mutations were shown to be disease-causing in EB (2), and 30 additional genes functionally related to epidermal adhesion or potential genetic modifiers (Table SI). NGS was performed using a custom Agilent Haloplex panel (Agilent, Santa Clara, CA, USA) and sequences were determined with an Illumina MiSeq (2 × 150 base pairs; Illumina, San Diego, CA, USA). Paired-end sequencing reads were aligned using Burrows-Wheeler Alignment tool (5) (version 0.7.15) against the human reference genome sequence GRCh37 (hg19). Variants were called using FreeBayes (6) (v1.1.0). ANNOVAR (ANNOtate VARiation) (7) was applied to annotate the function of genetic variants and to cross-reference the variants with databases. Only variants with a frequency of < 1% and homozygous absence in ExAC (8) v0.3 were considered. Mutations were confirmed by Sanger sequencing, and whenever possible, segregation was verified in the families (primer sequences available on request). Direct and indirect immunofluorescence were performed as described previously (9, 10). Fluorescein (FITC)-labelled antibodies used for direct immunofluorescence were anti-human IgG, IgA, IgM and C3c (Dako, Hamburg, Germany) at a dilution of 1:200, 1:50, 1:50 and 1:500. For IIF on salt-split skin, patient sera were diluted 1:10, secondary antibodies used were FITC-labelled anti-human IgG (Dako, Hamburg, Germany) at a dilution of 1:100. Commercial enzyme-linked immunoassay (ELISA) kits for the detection of NC16A domain of BP180- and bullous pemphigoid 230-kDa antigen (BP230)-specific antibodies (MBL) were used as per manufacturer’s protocol, with cut-off at 9 U/ml.
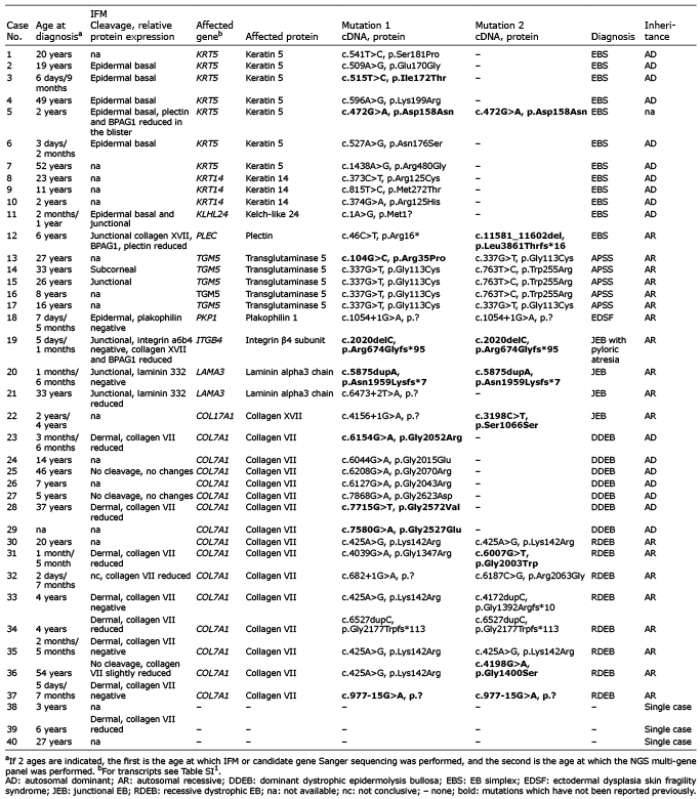
Analysis of a skin biopsy by IFM was performed in 25 cases, and correctly established the EB subtype in 19 of these (76%). In 36 cases (90%) the molecular patho-logy was completely elucidated by the targeted NGS multi-gene panel (Table I). In 3 cases, no mutations were disclosed: 2 had discrete acral blistering or peeling, and one was suspected to have dystrophic EB (cases 38–40 in Table I). Finally, in one case, only one known LAMA3 recessive mutation was disclosed.
Table I. Summary of the immunofluorescence mapping (IFM) and sequencing results
Twelve cases (1–12 in Table I) had basal EB simplex (30% of this cohort), 7 with mutations in KRT5, 3 in KRT14, 1 in KLHL24 and 1 in PLEC. Two KRT5 mutations have not been reported previously, the other mutations being recurrent. In case 5, the KRT5 mutation c.472G>A, p.Asp158Asn was identified in a homozygous state, but information on the phenotype of the patient and parents was not available in order to establish the inheritance pattern. Case 11 is a girl with congenital skin fragility, which improved soon after birth. At the age of 2 months, a skin biopsy was analysed showing basal and junctional cleavage. Subsequently, genes known to be associated with simplex and junctional EB were sequenced with the Sanger method, but yielded no evidence for disease-causing mutations. The implementation of the NGS multi-gene panel coincided with the discovery of KLHL24 as a new EB gene, and the recurrent mutation c.1A>G in the translation initiation codon of KLHL24 (11, 12) was disclosed, allowing the diagnosis. Case 12 is a 6-year-old boy with generalized skin blistering and nail dystrophies. Immunoreactivity for several components of the hemidesmosomes, such as collagen XVII, BPAG1, plectin was altered in the skin sections. Two mutations in a compound heterozygous state were found in the gene for plectin, PLEC. One of these, c.46C>T, p.Arg16*, located in exon 1a, was recently reported to cause a new autosomal recessive plectinopathy comprising EB simplex without muscular dystrophy (13).
Five cases (13–17) had TGM5 mutations and acral peeling syndrome, confirming the significant contribution of the mutations in this gene to the molecular patho-logy of EB simplex (27.7% in this cohort). Finally, in case 18, a homozygous PKP1 mutation was found, as predicted by IFM performed in the first days of life, establishing the diagnosis of ectodermal dysplasia-skin fragility syndrome.
Junctional EB was diagnosed in 4 cases (19–22 in Table I). In cases 19 and 20, IFM findings were confirmed by NGS, which disclosed homozygous ITGB4 or LAMA3 mutations, respectively. Case 21 is a 33-year-old woman with late onset skin fragility, in which a bullous autoimmune disorder was suspected, but excluded by direct immunofluorescence. Only one heterozygous LAMA3 mutation was identified, c.6473+2T>A, which was previously reported in patients with autosomal recessive severe generalized junctional EB (14). No pathogenic variants in other EB genes including LAMB3 or LAMC2 were disclosed. Nevertheless, reduction in the immunoreactivity for laminin 332 strongly supports the diagnosis of junctional EB.
Case 22 is a 4-year-old boy with mild acral skin fragility. Clinically, simplex or dystrophic EB were suspected, and mutations in the respective genes had been excluded before by Sanger sequencing. Multi-gene panel analysis showed that the only gene harbouring two heterozygous pathogenic variants was COL17A1, allowing the classification of the case as localized junctional EB. The identified mutations were c.4156+1G>A and c.3198C>T, p.Ser1066Ser. The latter is predicted to affect splicing (Mutation Taster: disease causing 0.99; Minor allele count A=0.00002/2 (ExAC), A=0.00008/1 (GO-ESP)), and was disclosed in 3 additional cases with junctional EB in our cohort.
COL7A1 mutations were found in 15 patients (cases 23–37 in Table I), being the most common cause of EB in this cohort (37.5%). Seven patients had dominant and 8 had recessive dystrophic EB. Most COL7A1 mutations were recurrent; to the best of our knowledge, 6 mutations were not reported previously. Identification of COL7A1 mutations was crucial in establishing the diagnosis of dystrophic EB in cases 28 and 36 at the ages of 37 and 54 years, respectively. Both women had late onset of skin fragility during adulthood, which has been considered to be acquired. Direct immunofluorescence clearly excluded the diagnosis of an autoimmune blistering disorder in case 36. Results were inconclusive in case 28, but skin blistering was refractory to immunosuppressive therapies. In this case we suspect a dominant dystrophic EB and a questionable immunoreactivity mimicking EB acquisita (15). In case 37, absence of collagen VII in the skin allowed the diagnosis of dystrophic EB by IFM in the first week of life, while the underlying intronic COL7A1 mutation, c.977-15G>A, might have been missed without information on the candidate gene.
This broad multi-gene panel was designed to also address the question as to whether additional variants, other than the disease-causing variants, might have a disease-modifying role in EB (16, 17). Indeed, beside the disease-causing variants, several variants were found in the other genes of the panel, in particular in PLEC (Table SII). Their interpretation, especially regarding modifying effects, is challenging. Based on these preliminary data, we believe that complex constellations of disease-causing mutations and genetic modifiers occur in individual situations. They should be suspected and carefully investigated if phenotypic variability occurs between members of the same family (16, 18), but are impossible to trace in a cohort of patients with heterogeneous background.
This EB gene-panel also delivered incidental findings. Two of the patients with EB simplex due to KRT5 or KRT14 mutations were also carriers of heterozygous recurrent pathogenic LAMB3 variants (c.1903C>T, p.Arg635* and c.628G>A,p.Glu210Lys, respectively), which is relevant for genetic counselling. A modifying role of these variants on the abnormal keratin intermediate filaments is unlikely.
This study evaluates the efficacy of a NGS multi-gene panel in the diagnosis of EB. The targeted NGS approach significantly expedited the diagnosis in cases with atypical clinical and IFM constellations, as reported elsewhere (19, 20). This approach conveniently covers all EB genes at once and the costs for NGS are decreasing. Still, this
method is not available/affordable in every country. Before initiation of this comprehensive analysis, patients must be informed regarding incidental findings and their delivery. The possible reasons why NGS fails in identifying mutations are either technical (i.e. DNA regions not covered by sequencing, large deletions, which cannot be properly detected), or conceptual (i.e. a gene not included in the panel, misdiagnosis, lack of precise clinical information). We believe that the second situation applies to cases 38–40 in this study. Mutations in other genes that are not included in this panel, or acquired disorders, may be responsible for mild acral blistering and peeling observed in cases 38 and 40, and for palmoplantar blistering and striate keratoderma in case 39.
It is noteworthy that IFM, an almost 40-year-old method (21), was able to deliver the correct classification of EB in more than 70% of cases. In this study, IFM has failed to solve the EB simplex cases due to KLHL24 (case 11) and PLEC (case 12) mutations, as well as one acral peeling syndrome case (case 15), because of the misleading, probably artificial junctional skin cleavage. In one recessive (case 36), and 2 dominant dystrophic EB cases (cases 25, 27), no skin cleavage was observed, and no anomaly of the immunoreactivity of collagen VII was found in the latter 2. Nevertheless, IFM proved successful in establishing the EB subtype in particular in neonates (22). It allows for fast diagnosis (the earliest IFM was performed at 2 days of age), which can aid the counselling of parents in the stressful situation of having an EB-affected newborn. IFM is also important in addressing the consequences of genetic variants of unknown significance on protein level. Finally, knowledge of the impact of the mutation on protein abundance indicated by IFM (i.e. complete absence/reduced/normal protein expression) is often a requisite for eligibility of patients for clinical trials or specific therapeutic interventions.
This study underscores the efficacy of the strategy of combining targeted NGS with IFM in resolving unusual phenotypes. It also suggests that, despite technological advances, careful clinical evaluation and deep phenotyping remain crucial factors for successfully establishing EB diagnosis.
We thank all clinicians who sent samples, the EB Center Freiburg, Käthe Thoma and Annegret Bedorf for excellent technical support. The work of CH was supported by Debra International.


 Click to show fullsize
Click to show fullsize