


1Institute of Human Genetics, University Hospital Magdeburg, Otto-von-Guericke University, Leipziger Str. 44, DE-39120 Magdeburg, 2Medical Faculty Carl Gustav Carus, Technical University, Dresden, Germany, 3Division of Human Genetics, Medical University of Innsbruck, Austria, and 4Deparment of Dermatology, Medical Center – University of Freiburg, Germany. E-mail: martin.zenker@med.ovgu.de
Accepted Jan 15, 2018; Epub ahead of print Jan 16, 2018
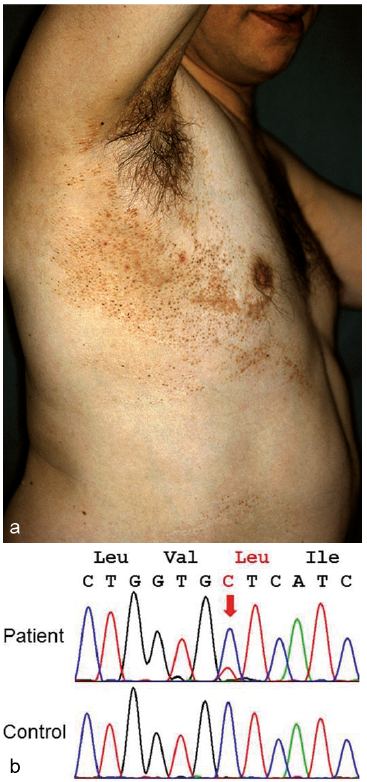
In 2008, we delineated a distinct sporadically occurring syndrome in the form of segmentally arranged basaloid follicular hamartomas with osseous, dental and cerebral anomalies (1). The reported patient was a 39-year-old man with multiple skin tumours that showed, in part, comedo-like plugging and were arranged in a unilateral, systematized pattern following Blaschko’s lines (Fig. 1a). The patient’s ipsilateral (right) leg was 22.5 cm shorter than the left leg. Rudimentary preaxial polydactyly was noted on the left hand and right foot. The right maxillary teeth were hypoplastic. Moreover, the right side of his chest was rather hairless, and linear areas of atrophoderma and hyperpigmentation involved the right arm and lower leg. Eight similar cases were found in the literature, and we argued that this disorder should not be conflated with mosaic forms of Gorlin syndrome (1). Subsequently, several similar cases were reported (2–6), and the name “Happle-Tinschert syndrome” (HTS) was proposed (2).
Fig. 1. Happle-Tinschert syndrome. (a) Unpublished photograph of multiple basaloid follicular hamartomas in the original case from 2008 (1). (b) Demonstration of the mosaic SMO mutation c.1234C>T (p.Leu412Phe) in lesional tissue from the patient.
In 2016, Khamaysi et al. (7) reported an activating SMO mutation in what they believed to represent a mosaic form of Gorlin syndrome. In a Letter to the Editor (8), we argued that their patient did not have “segmental basal cell naevus syndrome”, but HTS. Moreover, we suggested that HTS may turn out to be a clinical variant of Curry-Jones syndrome (CJS) that is likewise caused by postzygotic SMO mutations (9). In their reply, however, Khamaysi et al. (10) upheld their opinion that the presented case belonged to the spectrum of Gorlin syndrome because the patient “displayed diagnostic criteria for Gorlin syndrome but not for HTS or CJS”.
Meanwhile, we had the opportunity to analyse the SMO gene in DNA from a native cryoconservated tissue sample obtained from a skin area with basaloid follicular hamartomas of the original HTS patient reported in 2008 (1). Genotyping was performed by standard Sanger sequencing. As demonstrated in Fig. 1b, we could identify the sequence change c.1234C>T in the DNA sample. The fraction of mutated DNA was calculated to be 20–30%, thus suggesting a mosaic status. The observed mutation predicts the missense change p.Leu412Phe in the SMO protein and is the known recurrent SMO mutation that had been found in CJS (9) and in the patient reported by Khamaysi et al. (7). These findings corroborate our previous hypothesis that HTS is indeed caused by mosaic SMO mutations and represents a variant of CJS. We therefore postulate that the phenotype reported by Khamaysi et al. (7) belongs to this spectrum of sporadically occurring mosaic SMO disorders.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize