


Takuya Miyagawa, Makoto Sugaya*, Asako Okada, Yoshihide Asano, Takafumi Kadono and Shinichi Sato
Department of Dermatology, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, 113-8655, Japan. *E-mail: sugayam-der@h.u-tokyo.ac.jp
Accepted Mar 12, 2014; Epub ahead of print Mar 25, 2014
Anaplastic large cell lymphoma (ALCL) is characterized by cohesive infiltration of large atypical lymphocytes with abundant cytoplasm. According to the WHO classification, more than 75% of ALCL tumour cells express CD30 (1). CD3 may be absent or expressed at lower levels than in other cutaneous T-cell lymphomas and reactive T cells (2). Cutaneous lesions usually present as erythematous to violaceous tumours that frequently show ulcerations. Small nodules, subcutaneous tumours, pedunculated tumours, indurated erythematous plaques, lichen planus-like skin lesions, and erythroderma are also reported (3–5). In some cases, cutaneous ALCL lesions are misdiagnosed as different skin diseases. Pseudocarcinomatous epidermal hyperplasia is frequently seen in primary cutaneous ALCL, which is clinically similar to squamous cell carcinoma (6). Papules and plaques simulating morphoea or ulcerated lesions resembling pyoderma gangrenosum have also been described (7). We report here a rare case of ALCL mimicking furunculosis characterized by an abundant infiltration of neutrophils and high interleukin-8 expression by tumour cells.
CASE REPORT
A 57-year-old Japanese man presented at our department with multiple tumours in both thighs and buttocks. He had received 4 courses of ABVD therapy (doxorubicin (adriamycin), bleomycin, vinblastine, dacarbazine) and 36 Gy focal irradiation in the right inguinal area following a diagnosis of Hodgkin’s disease 7 years earlier. Complete remission was achieved with this treatment. Two years after the chemotherapy, he developed lymphoedema of the right leg, which gradually worsened and extended to the left leg. He received bilateral lymphaticovenular anastomosis one month before visiting our department, but lymphoedema and fever above 38°C persisted. He began to develop multiple red-to-violaceous thumb-sized tumours and plaques in both thighs and buttocks (Fig. 1a). Some tumours and plaques easily became ulcerated. He and his family had no other medical history. A skin biopsy specimen showed abundant extravasation of erythrocytes and infiltration of lymphocytes and neutrophils in the dermis. Some epithelioid cells with large nuclei were also scattered in the dermis, which stained positively for Ki-67. Staining for CD31 revealed an increased number of vessels in the dermis. The epithelioid cells were negative for leukocyte common antigen (LCA). Laboratory findings on admission showed the following values: leukocytes, 15.4 × 109/l (normal 3.5–9.2 × 109/l) with 82% neutrophils; erythrocytes, 359 × 1010/l (normal 420–554 × 1010/l); platelets, 61.2 × 1010/l (normal 15.5–36.5 × 1010/l); lactate dehydrogenase (LDH), 158 IU/l (normal 125–237 IU/l); C-reactive protein, 17.47 mg/dl (normal: 0.0–0.3 mg/dl); and soluble interleukin-2 receptor (sIL-2R), 3,348 U/ml (normal: 167–497 U/ml). F-18 fluorodeoxyglucose (FDG) positron emission tomography scans showed multiple regions of abnormally increased FDG uptake in his left inguinal and right axillary regions, the middle of the abdominal cavity, and the left supraclavicular fossa. Based on these clinical and pathological findings, our first diagnosis was angiosarcoma associated with chronic lymphoedema (Stewart-Treves syndrome). Treatments with bi-weekly docetaxel (30 mg/m2) and recombinant interleukin (IL)-2 (35 × 104 units) were started, but the tumours mimicking furunculosis continued to develop on the right thigh and buttocks (Fig. 1b). His past history of lymphoma, frequent ulceration of the tumours, continuous high fever, leukocytosis, and unresponsiveness to docetaxel and IL-2 prompted re-biopsy. The new biopsy specimen showed infiltration of large lymphoid cells with marked nuclear atypia and sheets of neutrophils (Fig. 2a). Immunohistochemistry revealed that the atypical cells were positive for CD30 (Fig. 2b), CD4, granzyme B, perforin, and negative for CD8, anaplastic lymphoma kinase (ALK), Pax-5, epithelial membrane antigen (EMA) and LCA. The lymphocytes present in the first biopsy showed the same immunophenotype. The patient was diagnosed with ALCL and treated with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) therapy. The tumours reduced in size after the first course of the therapy, but they did not respond well to the second course. The patient passed away due to sepsis 2 months after the final diagnosis.
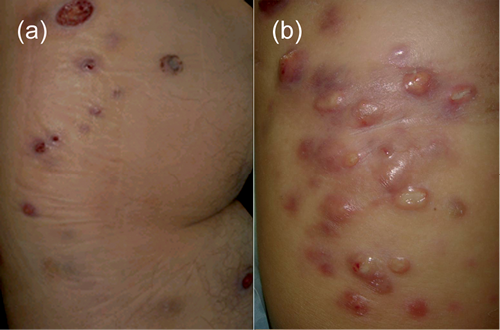
Fig. 1. (a) Erythematous to violaceous thumb-sized tumours and plaques in the left thigh and buttocks on admission. (b) Newly-developed tumours on the right thigh and buttocks mimicking furunculosis, after chemotherapy with docetaxel and recombinant interleukin (IL)-2.
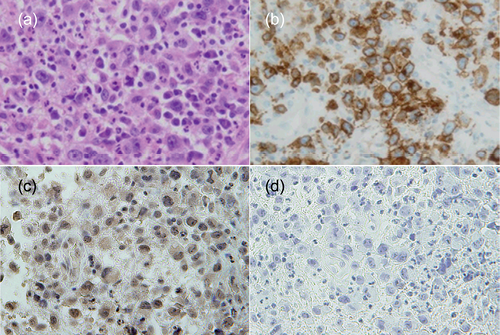
Fig. 2. (a) Histopathology showing infiltration of large lymphoid cells with marked nuclear atypia and abundant neutrophils (haematoxylin–eosin stain; original magnification: ×600). (b) Immunohistochemical staining of atypical large lymphocytes for CD30 (original magnification: ×600). (c and d) Immunohistochemical staining of (c) atypical large lymphocytes for interleukin-8 and (d) isotype control (original magnification: ×600).
DISCUSSION
Our final diagnosis of this case was ALCL, ALK-negative. The biopsy specimen obtained 7 years before was not available and we could not confirm whether lymphoma in the right inguinal area was from the same clone. Erythematous to violaceous tumours and plaques secondary to chronic lymphoedema, massive extravasation of erythrocytes, and an increased number of vessels led to an initial misdiagnosis of Stewart-Treves syndrome. An abundant infiltration of inflammatory cells and negativity for LCA expression by the atypical epithelioid cells made it difficult to diagnose lymphoma. In certain cases of malignancy, including lymphoma, tumour cells sometimes lose surface expression of lineage-specific markers (6, 8), as was the case described here.
There are several histological subtypes of ALCL. The distinct variants of ALCL in the WHO classification are the common (classic), lymphohistiocytic, small cell, and Hodgkin-like variants (1). In addition, other disease variants such as giant cell-rich, sarcomatoid, “signet ring”-like, eosinophil-rich, and neutrophil-rich types, have been recognized (9–14). The neutrophil-rich form was first described by Mann et al. in 1995 (14). It was characterized by a significant neutrophilic infiltrate accompanied with peripheral neutrophilia. It is noteworthy that neutrophil-rich ALCL may present diagnostic difficulties because many inflammatory leukocytes can mask a small number of tumour cells. Dense infiltration of neutrophils is often associated with bacterial and fungal infections, Sweet’s syndrome, and pyoderma gangrenosum. Interestingly, we performed immunostaining for interleukin-8 (LifeSpan BioSciences, Inc., Seattle, WA, USA), a potent neutrophil chemotactic factor, and found that tumour cells expressed this cytokine (Fig. 2c and d). This may explain why the tumour contained such large numbers of neutrophils. Similarly, a patient with Sézary syndrome and disseminated pustulosis was previously reported, where tumour cells expressed high levels of interleukin-8 (15). The important lesson from this report is that neutrophil-rich ALCL should be considered in patients with cutaneous tumours displaying neutrophil-related features, such as pustules, high fever, neutrophilia and an unusual number of neutrophils in cutaneous lesions upon histopathological analysis, among other, mainly infectious or reactive conditions.
ACKNOWLEDGEMENT
The authors would like to thank Dr Andrew Blauvelt of the Oregon Medical Research Center in Portland, Oregon, USA, for critical review of this manuscript.
REFERENCES

