


Dimitra Kiritsi1#, Laura Huilaja2,3#, Claus-Werner Franzke1, Nina Kokkonen2,3, Chiara Pazzagli1,4, Agnes Schwieger-Briel1, Markku Larmas5, Leena Bruckner-Tuderman1, Cristina Has1*, and Kaisa Tasanen2,3
Departments of Dermatology, 1Medical Center-University of Freiburg, Hauptstraße 7, DE-79104 Freiburg, Germany, 2Oulu Center for Cell-Matrix Research, University of Oulu, 3Medical Research Center, Oulu University Hospital, Oulu, Finland, 4Spemann Graduate School of Biology and Medicine (SGBM), Albert Ludwigs University Freiburg, Freiburg, Germany and 5Institute of Dentistry, University of Oulu, Oulu, Finland. *E-mail: cristina.has@uniklinik-freiburg.de
#These authors contributed equally to this paper and should be considered as first authors.
Accepted Feb 16, 2015; Epub ahead of print Feb 24, 2015
Laminin-332, a heterotrimeric glycoprotein consisting of laminin α3, β3 and γ2 chains, is a major component of the cutaneous basement membrane, connecting keratinocytes to the underlying dermis (1). Its biological relevance is highlighted by its link to human pathologies, e.g. inherited and acquired skin fragility disorders, and by mouse models (2). In humans, more than half (57%) of the mutations in the genes coding for the laminin-332 chains (LAMA3, LAMB3 and LAMC2) lead to premature termination codons (PTC), and presumably to loss of expression of the corresponding polypeptides (HGMD professional https://portal.biobase-international.com). Such biallelic PTC mutations are associated with severe generalised junctional epidermolysis bullosa (JEB) (formerly known as Herlitz) which manifests with congenital generalised skin and mucosal blistering and early demise within the first years of life due to respiratory failure, failure to thrive, anaemia and infections (2–5). In most cases the laminin β3 chain is affected, with the mutation p.R635* accounting for 40–60% of the mutant alleles (6). Milder clinical features, such as generalised intermediate, localised or late onset skin fragility have been associated with mutations in the genes for laminin-332 chains, collagen XVII or integrin α6β4. JEB patients are often affected by abnormal dental development and generalised or focal enamel hypoplasia, meaning amelogenesis imperfecta.
Here, we unravel the molecular mechanisms underlying mild cutaneous fragility in 2 patients, who were compound heterozygous for the recurrent mutation p.R635* of the laminin β3 chain, and 2 distinct splice site mutations. The latter allowed expression of truncated laminin β3 chains, which prevented a dramatic outcome.
CASE REPORTS (for Methods see Appendix S11)
Case 1. A 38-year-old Finnish male, developed skin blistering shortly after birth. Currently, blistering occurs most often on face, feet, legs, dorsal hands and arms, and is more pronounced during the summer time (Fig. S1A–C1). Toe nails are dystrophic, but finger nails normal (Fig. S1B and C1). As a teenager, all his permanent teeth had to be restored with crowns due to severe amelogenesis imperfecta. JEB was diagnosed at the age of 33 by transmission electron microscopy of a skin sample, which demonstrated cleavage within the lamina lucida, rudimentary hemidesmosomes and altered anchoring fibrils (not shown).
Case 2. A 16-year-old German female, had also suffered from congenital skin blistering. Immunofluorescence mapping of a postnatal skin biopsy specimen with antibodies to the cutaneous basement membrane proteins (7) showed a junctional split of the skin and strongly reduced staining for the laminin-332 chains. These findings were compatible with the diagnosis of severe JEB and a poor prognosis. Unexpectedly, skin fragility improved with age and currently, blisters occur almost exclusively on the hands and feet (Fig. S1D and E1). In addition, she had high propensity to dental attrition due to amelogenesis imperfecta (Fig. S1F1). Both patients have normal hair and no mucosal involvement.
To determine the molecular basis of the disease, LAMB3 mutation analysis was performed in both index cases and their parents. In Case 1, 2 heterozygous mutations were found, c.1903C>T; p.R635*, and c.3052-1G>A, which disturbs the acceptor splice site of exon 21 (Fig. 1A). The mother was heterozygous for c.1903C>T, p.R635*, and the father for c.3052-1G>A. In Case 2, c.1903C>T; p.R635* was disclosed, and c.1288+1G>T at the donor splice site of exon 10 (Fig. 1A). To the best of our knowledge, c.3052-1G>A and c.1288+1G>T have not been previously reported (HGMD Professional 2014.2), but they affect highly conserved positions and are predicted to disrupt the respective splice sites. To uncover the consequence of these mutations, RNA studies were performed. In Case 1, c.3052-1G>A resulted in a transcript with in-frame skipping of the first 6 nucleotides of exon 21 in about 40% of the analysed clones (n = 16) (Fig. 1B). Prediction with NetGene2 shows that c.3052-1G>A alters the normal acceptor splice site (confidence decreases from 0.76 to 0.34), resulting in the usage of the neighbouring cryptic splice site, 6 base pairs downstream. On protein level, this is predicted to result in the deletion of 2 amino acids, valine and glutamine at positions 1018 and 1019, within a large coiled-coil domain of the laminin β3 polypeptide (p.V1018_Q1019del). In Case 2, c.1288+1G>T resulted in in-frame skipping of exon 10, which is predicted to result in the deletion of 63 amino acids, from arginine 315 to serine 378 (p.R315_S378del), representing the entire EGF-like domain 2 (LE 2) (8).
To address whether the predicted truncated polypeptides were translated and secreted, immunoblot and immunostainings were employed. In the skin of both patients, the staining of the laminin β3 chain was reduced (Fig. 1C). The positive immunostaining of all laminin-332 chains (Fig. S2A1) in both patients suggested that the laminin β3 molecules truncation did not prevent heterotrimerisation, secretion and assembly of laminin-332 into the basement membrane. The semi-quantitative evaluation of the laminin β3 staining revealed reduced signals, corresponding to approximately 50% of normal levels in case 1, and about 13% in case 2 (Fig. 1C). In case 1 the deletion of 2 amino acids was not detectable in immunoblots, whereas in case 2 truncation of the polypeptide was reflected by the presence of a band migrating below the normal 140 kDa laminin β3 chain in the control sample. The truncated laminin β3 polypeptide with LE2 deletion was mainly detected in keratinocyte lysate indicating intracellular accumulation (Fig. S2B1).
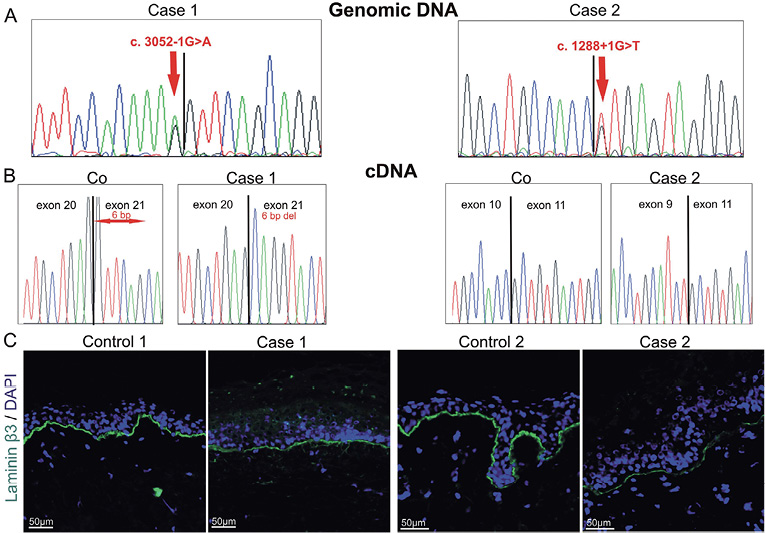
Fig. 1. LAMB3 splice site mutations c.3052-1G>A and c.1288+1G>T and their consequences. A. Partial genomic DNA sequences indicating the mutations c.3052-1G>A and c.1288+1G>T (red arrows). B. Partial cDNA sequences demonstrating the splicing defects resulting from the splice site mutations: deletion of 6 base pairs in case 1 (6 bp del) and skipping of exon 10 in case 2; Co: control sequences. C. Immunofluorescence staining with antibodies to the laminin β3chain in skin from control and cases 1 and 2.
To get insights into the impact of p.V1018_Q1019del and p.R315_S378del on the laminin β3 polypeptide, secondary structure prediction was performed using SSpro 4.5 software (http://scratch.proteomics.ics.uci.edu/explanation.html) (9). In neither case significant alterations were seen, as compared to the wild type laminin β3 chain (not shown). The coiled-coil structure of the laminin β3 chain was assessed with COILS software (http://embnet.vital-it.ch/software/COILS_form.html) (10). One coiled-coil region was predicted to be disturbed from the 2 amino acid deletion p.V1018_Q1019del in the C-terminus of the laminin β3 chain (Fig. S31), whereas no significant changes were predicted in the case of deletion of the LE2 domain, p.R315_S378del.
DISCUSSION
The genotype-phenotype correlations in the present cases reveal that LAMB3 splice site mutations allowing in frame skipping and expression of truncated laminin β3 chains are associated with mild skin fragility, amelogenesis imperfecta and nail dystrophy. The fact that both cases have minor skin fragility and normal life expectancy, suggests that small amounts of partially functional laminin-332 are sufficient for substantial cutaneous stability and prevent mucosal involvement. Enamel formation is disturbed since laminin-332 is expressed during tooth development by ameloblasts, and required for cell adhesion of the odontogenic epithelium and normal mineralisation of enamel (11). Alongside information on the laminin-332 amount, which is critical for dermal–epidermal adhesion, these findings shed light on the functions of laminin β3 domains. The deletion p.V1018_Q1019del in the C-terminus of the laminin β3 chain, which is critical for laminin chain assembly (12), allows secretion of laminin-332, but is predicted to alter its coiled-coil structure and consequently reduce the laminin-332 heterotrimer stability. The role of the laminin β3 LE domains has remained elusive (13). Our findings suggest that deletion of LE2 interferes with laminin-332 secretion, probably due to protein folding defects (11).
Our findings have therapeutic relevance. A few similar cases (4–6, 14, 15) were reported before; in particular, in an Italian patient, the homozygous frameshift LAMB3 mutation illegitimated splicing, resulting in shortened, but functional laminin β3 polypeptides and therefore only mild disease (4). These genotype-phenotype correlations suggest that in frame skipping of exons harbouring mutations leading to premature termination codons may be a therapeutic alternative in JEB with LAMB3 mutations.
Acknowledgements
We thank the patients and their families for their cooperation in this research project. The excellent technical support by Vera Morand, Ioannis Athanasiou, Anja Mattila, Margit Schubert and Kaethe Thoma is gratefully acknowledged. This work was supported by the grant SFB850-B6 to LBT and CWF, and the “Theodor-Nasemann Scholarship” from Galderma Förderkreis and the German Research Foundation DFG [grant 1795/1-1] to DK and the grants from the Academy of Finland and the Oulu University Hospital to KT. Further, this study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School). The contribution of Prof. Jürgen Kohlhase (Center for Human Genetics Freiburg) is acknowledged.

1http://www.medicaljournals.se/acta/content/?doi=10.2340/00015555-2073
REFERENCES


