


Min Soo Jang, Dong Young Kang, Jong Bin Park, Sang Hwa Han, Kang Hoon Lee, Joon Hee Kim, Jae Won Ko and Kee Suck Suh*
Department of Dermatology, Kosin University College of Medicine, 262 Gamcheon-ro, Seo-gu, Busan, 602-702, South Korea. *E-mail: ksderm98@unitel.co.kr
Accepted Jun 3, 2015; Epub ahead of print Jun 10, 2015
Mycosis fungoides (MF) is the most common form of cutaneous T-cell lymphoma (CTCL) and is characterized by the clonal expansion of skin-homing T lymphocytes. Various clinical and histological variants of MF have been reported (1).
Ichthyosiform MF (IMF) is not as well-known as other variants of MF. Clinical and pathological findings of previous studies are inconsistent, which has made it difficult to define this variant. We report here the clinical and histopathological features of patients with IMF who visited our hospital.
MATERIALS AND METHODS (see Appendix S11)
RESULTS
Clinical findings (Fig. 1 and Tables SI and SII1). Ten patients with IMF (7 males and 3 females) were studied (mean age 32 years (range 8–63); 3 were ≤ 20 years of age). The duration of disease before diagnosis ranged from 2 to 20 years, with a mean of 6.6 years. Five patients had pruritus, 3 of whom reported severe pruritus. Seven patients had scattered ichthyosiform lesions all over their body, and one of them had these symptoms on the face and scalp; the 3 remaining patients had lesions only on the trunk and extremities. In 4 patients (Nos. 2, 3, 9 and 10) presence of ichthyosiform lesions was the sole manifestation of MF. Four patients had conventional MF lesions; 2 had ichthyosiform lesions that appeared concurrently (Nos. 1 and 4) and the remaining 2 (Nos. 6 and 8) experienced patches or plaques first, and then the ichthyosiform lesions appeared 5 and 4 years later. One of the 2 remaining patients (No. 8) had a folliculotropic lesion and primary cutaneous anaplastic large cell lymphoma 1 and 3 years after the ichthyosiform lesions developed, respectively. A hypopigmented lesion (No. 7) and an ulcerative lesion (No. 5) also developed. According to TNM classification, stage IB was confirmed in 9 patients, and IIA in 1 patient.
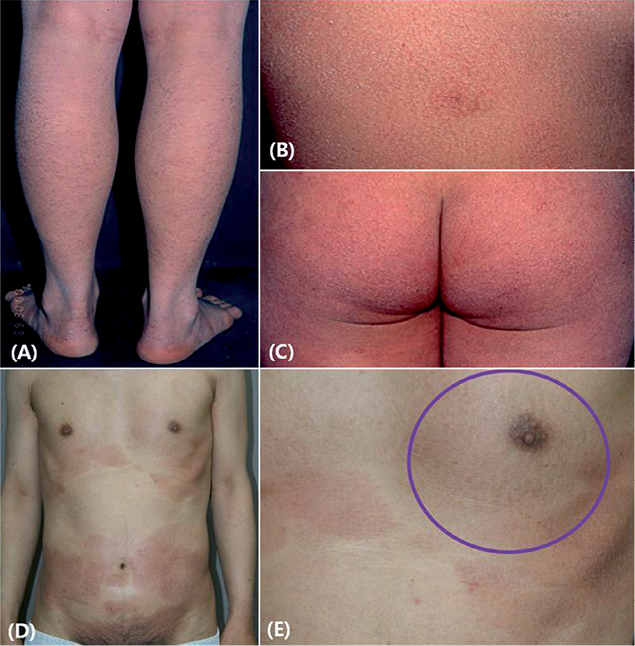
Fig. 1. (A–C) Ichthyosiform eruption as the sole manifestation of ichthyosiform mycosis fungoides (MF). Ichthyosiform eruption on various sites (patient 3). (D, E) Ichthyosiform eruption (circle) in conjunction with the classic types of MF (patient 6).
Psoralen plus ultraviolet A (PUVA) only (n = 4), acitretin and PUVA (RePUVA) (n = 4), and narrowband UVB (NBUVB) (n = 2) were given as initial treatments. Partial responses were observed in all of the patients treated with PUVA only. All patients with RePUVA also showed partial responses and methotrexate, ultraviolet A-1 (UVA-1), acitretin, and topical steroids were used as second or third treatments. Among 2 patients who underwent NBUVB as an initial treatment, one responded well to NBUVB; however, the other patient did not respond and the condition progressed to folliculotropic MF. The follow-up period after diagnoses ranged from 1–18 years, with a mean of 6.7 years. None of the patients showed progression toward a tumour or lymph node stage.
Histological, immunohistochemical and molecular findings (Figs S1, S2, and Table SIII1). In all 10 patients, biopsy of ichthyosiform lesions showed both MF and ichthyosis vulgaris-like features. In the epidermis, epidermotropism and “haloed” lymphocytes were observed in all 10 cases, and parakeratosis and Pautrier’s microabscess were observed in 5 cases each. In the dermis, coarse collagen bundles were observed in the dermal papilla in all 10 cases. Compact orthokeratosis and thinning or absence of granular layer were observed in all patients, and folliculotropism (No. 8) and fibrinoid necrosis (No. 5) in one case each.
Four cases had a CD4/CD8 ratio greater than 1; in 4 cases the CD4/CD8 ratio was 1, and 2 cases had a CD4/CD8 ratio of less than 1. In 6 of 10 cases, CD7 expression was found in less than 10% of the infiltrated cells.
Assessment of TCRγ gene rearrangement was confirmed monoclonality in 6 cases (60%).
DISCUSSION
In 1984, Aram (4) first reported ichthyosiform lesions as a clinical manifestation of MF. In 1996, Kutting et al. (5) described a 25-year-old patient with an ichthyosiform lesion that had both MF and ichthyosis vulgaris-like histological findings.
In the current study, patients’ mean age was 32 years, younger than previously reported for both MF (55–60 years) and IMF (51–59 years) (6–9).
IMF can present with a variety of clinical and histological manifestations (6). In this study, the patients can be divided into 3 groups: those with ichthyosiform lesions only (patients 2, 3, 9 and 10); those with ichthyosiform lesions accompanied by classical MF; and those with ichthyosiform lesions accompanied by other MF variants, such as hypopigmented or ulcerative lesions. Previous studies have reported IMF accompanied by classical MF or follicular MF (6, 9), but there have been no reports of IMF accompanied by primary cutaneous anaplastic large cell lymphoma.
All patients in this study had histological findings that are expected in early MF, together with compact orthokeratosis and thinning or abscence of granular layers suggestive of ichthyosis vulgaris. Immunohistochemical testing revealed more CD8+ cells than CD4+ cells, and 6 cases showed monoclonality in TCR-γ gene rearrangement, similar to that seen in previous studies (6). Interpretations of clinical symptoms, histological results, and immunohistochemical staining may be needed to make appropriate diagnoses (10).
In conclusion, IMF can be considered as a rare type of early MF with a comparatively favorable prognosis, which is common in young patients. When examining patients with ichthyosiform lesions, careful evaluation including skin biopsy is necessary to rule out the possibility of IMF.

1http://www.medicaljournals.se/acta/content/?doi=10.2340/00015555-2159
REFERENCES


