


Departments of 1Dermatology, and 2Radiology, The First Hospital of China Medical University, 155 North Nanjing Street, Shenyang 110001, China. E-mail: cmuxt@126.com
Acrodermatitis continua of Hallopeau (ACH) is a rare chronic inflammatory skin disorder, characterized by recurrent sterile pustular eruptions, which first affects the extremities of digits and leads to onychodystrophy, anonychia and osteolysis of the distal phalanx (1). We report here a case of ACH lasting for 30 years, presenting with multiple granuloma-like vegetations and osteolysis of the digits, which was refractory to mycophenolate mofetil (MMF), but controlled successfully by acitretin.
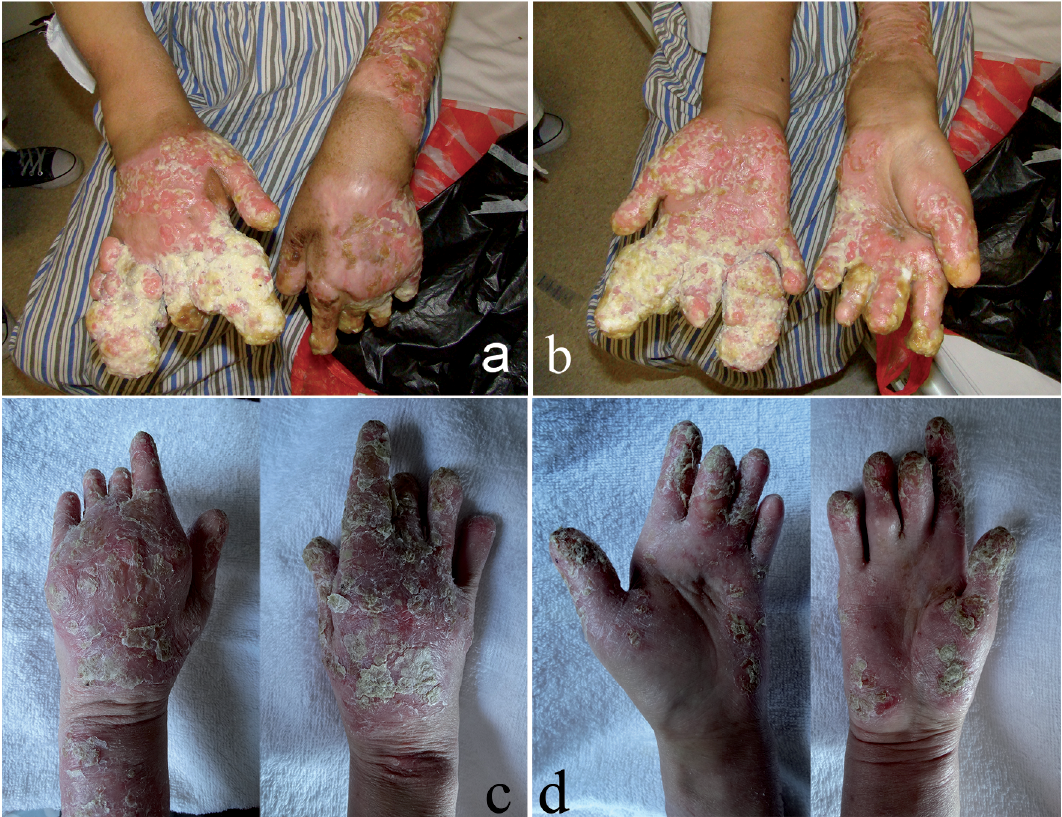
A 54-year-old Chinese woman presented with a 30-year history of ACH. She had initially developed pustules on the tip of the middle finger of the left hand. Subsequent repeated episodes of painful pustular eruptions involved more digits (Fig. S1a, c, d) accompanied by high fever and generalized pustules (Fig. S1b). She had no familial history of psoriasis. Oral cyclosporine, 5 mg/kg/day, was effective, but had been discontinued 20 years earlier due to impaired renal function. She had been taking MMF, 2.0–3.0 g daily, since then. Multiple vegetations had occurred 12 years previously (Fig. S1e, f). Topical antibiotics, antifungal agents, corticosteroids, oral tripterygium glycosides and dapsone had no effect. Physical examination revealed complete anonychia of both hands, absence of 6 distal digits, multiple granuloma-like vegetations of the digits covered by purulent exudates (Fig. 1a, b), fissured tongue, erythematous plaques and pustules on the left forearm and back. X-ray digital radiography showed a complete absence of 6 terminal phalanges, diffuse osteoporosis of phalanges, metacarpi, carpi, ulnae and radii, with prominent swelling of soft tissues (Fig. S1g). Histopathology of the vegetative lesions showed irregular epidermal hyperplasia, mild spongiosis with sparse neutrophils in the epidermis, dermal oedema, diffuse dermal infiltrates comprised primarily of neutrophils, and dilated small vessels filled with neutrophils (Fig. S2a). Laboratory tests showed total white blood cell (WBC) count 15.51 × 109/l (normal 3.50–9.50) with 90% neutrophils, red blood cell (RBC) count 2.76 × 1012/l (3.80–5.10), haemoglobin 63 g/l (115–150), serum iron 2.5 μmol/l (6.6–26.0), C-reactive protein (CRP) 48.6 mg/l (0–8), procalcitonin 0.13 mg/ml (0–0.05), albumin 22 g/l (40–55), creatinine 296 μmol/l (45–84). Repeated cultures from pustules and blood for bacteria and fungi were negative. Bacterial smears of purulent exudates of both hands revealed Gram-positive cocci and Gram-negative bacilli. Swab cultures of exudates revealed growth of diphtheroid bacillus. Serum anti-streptolysin antibodies, rheumatoid factors, and antinuclear antibodies were negative. HLA-B27 antigen was negative. Serum antibodies against human immunodeficiency virus, and hepatitis A, B, C, E viruses were negative. Serum indirect immunofluorescence (IIF), anti-desmoglein (Dsg) 1, anti-Dsg 3, anti-bullous pemphigoid (BP) 180 and anti-BP230 antibodies (enzyme-linked immunoassay (ELISA) kits from MBL, Nagoya, Japan) were all negative.
Fig. 1. Clinical presentation of the proband before and after treatment. (a, b) Absence of 6 distal phalanges, multiple granuloma-like vegetations. (c, d) Granuloma-like vegetations resolved completely after 18 months.
After obtaining written informed consent, the entire coding regions of IL36RN, including the exon/intron boundaries, were sequenced using genome DNA samples from the patient and her father, as described previously (2). Sequencing analysis revealed a homozygous splice-site mutation, c.115+6T>C in the IL36RN gene of the patient (Fig. S2b) and a heterozygous mutation, c.115+6T>C in the IL36RN gene of her father (Fig. S1h). IL36RN mRNA expression in cutaneous epithelial cells was detected by reverse-transcriptase PCR using total RNA extracted from plucked hairs of the patients and normal controls, as described previously (3). The complementary DNA fragments from exons 2 to 4 were amplified and sequenced (Fig. S3) showing complete skipping of exon 3 of IL36RN (Fig. S4).
ACH, cutaneous bacterial infection, renal insufficiency, iron deficiency anaemia and hypoalbuminaemia were diagnosed. Intravenous moxifloxacin, 400 mg daily (for 5 days, followed by intravenous meropenem, 1.0 g twice daily for 10 days, adjusted according to the drug sensitivity test of bacterial cultures), oral MMF, 1.0 g twice daily, calcium dobesilate, 500 mg thrice daily, ketosteril, 4 tablets thrice daily, sodium bicarbonate, 1 g thrice daily, and topical fusidic acid thrice daily were administered. Leukocyte-depleted RBC, 2 U/day, was transfused for 3 days. Fever subsided at day 11, but new pustules persisted. Oral acitretin, 30 mg/day, was added. WBC count returned to normal and haemoglobin was 70 g/l at day 21. The patient was discharged at day 24. After 18 months, granuloma-like vegetations resolved completely, but erythematous plaques with scales persisted (Fig. 1c, d). The patient remained well on acitretin, 20 mg, and MMF, 2.0 g, daily.
ACH is thought to be a variant of pustular psoriasis with deficiency of interleukin (IL)-36 receptor antagonist (DITRA) due to (i) their similar histopathological features, (ii) the evidence that many cases of ACH progressed into generalized pustular psoriasis (GPP), and (iii) that both ACH and GPP can be caused by mutations in the IL36RN gene, which encodes interleukin-36 receptor antagonist (IL-36Ra) (2). To date, 15 mutations in the IL36RN gene have been reported in GPP cases from Tunisian, European, Japanese, Malaysian and Chinese populations (2, 4, 5). The homozygous c.115+6T>C mutation found in this study, leads to a frame-shift and an immediate premature termination codon in IL36RN and appears to be a mutation hot-spot in Asian patients with GPP (6, 7), and the c.338C>T mutation (p.Ser113Leu) is the most prevalent allele mutation in European patients with GPP (8, 9).
IL-36 cytokine IL-36γ and IL-36 genes IL36A, IL36B, IL36G and IL36RN are overexpressed in the lesional skin of plaque psoriasis (10). Nevertheless, a recent European cohort study of 251 patients with palmoplantar pustular psoriasis (PPP), has shown it to be associated with missense variants in CARD14, but not with loss-of-function mutations in IL36RN (11). Moreover, variations in genes of IL-19 subfamily cytokines and the ATG16L1 gene of the autophagy pathway may also influence the susceptibility of PPP (12, 13). The interaction of multiple susceptible genes may cause wide variation in phenotype in different psoriatic patients with the same IL36RN mutation.
ACH with granuloma-like vegetation has not been reported. In 1990, Miyagawa et al. (14) reported a 17-year-old Japanese boy with an acral granulomatous dermatosis. They did not diagnose the case as ACH because the patient had no primary pustular lesions in the fingers and the histopathology did not show the formation of Kogoj’s pustules in the epidermis. The diagnosis of ACH in our case was based on typical clinical manifestations, chronic recurrent duration, and the homozygous c.115+6T>C mutation in IL36RN. The coexistence of an autoimmune bullous disease was ruled out by negative IIF, anti-Dsg1, 3 and anti-BP180, 230 antibodies.
In this case, granuloma-like vegetations occurred gradually during MMF therapy and responded dramatically to acitretin.
This work was supported by the National Natural Science Fund (81470142) and the Program for New Century Excellent Talents in University (NCET-10-0905) of the Ministry of Education, China.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize