


1Department of Dermatology, Nippon Medical School, 1-1-5 Sendagi, Bunkyo-ku, Tokyo 113-8603, 2Department of Dermatology & Plastic Surgery, Akita University Graduate School of Medicine, Akita, and 3Department of Dermatology, Yamagata University Faculty of Medicine, Yamagata, Japan. E-mail: sae777@nms.ac.jp
Accepted Jun 15, 2016; Epub ahead of print Jun 28, 2016
Oculocutaneous albinism (OCA) is a group of autosomal recessive disorders characterized by defective melanin biosynthesis, which results in congenital hypopigmentation of the hair, skin, and eyes despite a normal melanocyte number. Co-occurrence of malignant melanoma (MM) is considered rare in patients with OCA, compared with that of non-melanoma skin cancer, such as squamous cell carcinoma (SCC) and basal cell carcinoma (BCC). Although around half of cases of MM in patients with OCA have been reported to be melanotic, the mechanisms of developing pigmented MM remains unclear. We report here a patient with OCA type 4 (OCA4) who presented with a melanotic MM, in which tumour cells showed high expression of microphthalmia-associated transcription factor (MITF).
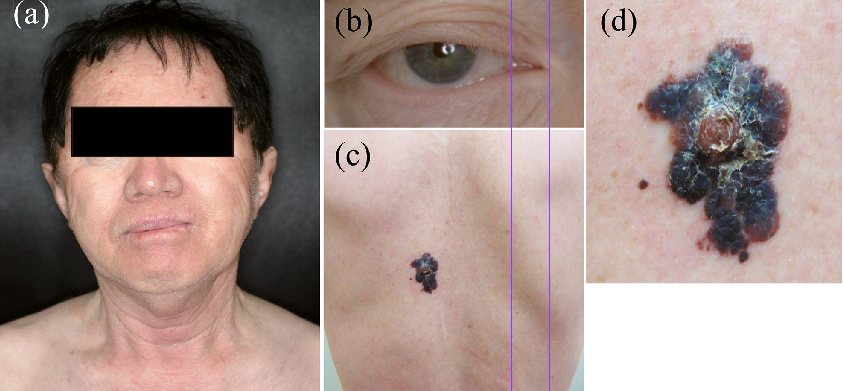
A 66-year-old Japanese man presented with a 6-month history of an enlarging black nodule on his back. Since birth, he had exhibited light yellow hair and hypomelanotic eyes and skin. He recalled experiencing sunburn during childhood, but had avoided sun exposure in older years. His parents were not consanguineous or affected by albinism, but his sister shared the same physical characteristics. On physical examination, the patient had hypomelanotic skin, gray-white irides, and bilateral horizontal nystagmus. His hair was dyed black. On his back was a black, firm, well-defined, irregularly-shaped tumour, 50×25 mm in size (Fig. 1). Systemic and laboratory examinations were normal. A melanoma was suspected, and excisional biopsy of the lesion was taken with 0.5-cm margins.
Fig. 1. Clinical features. (a) Patient with hypomelanotic skin and dyed black hair. Several painful oedematous nodules with blisters on the face at first visit. (b) Grey-white irides. (c) A black, firm, well-defined, irregularly-shaped tumour on the back. (d) Size 50×25 mm, with an erythematous plaque within the tumour.
Histological examination revealed a superficial spreading MM (Fig. S1 a, b). Melanin pigments were detected in some of these cells. Breslow thickness was 4.2 mm (Clark’s level IV). The patient subsequently underwent local re-excision with additional 2-cm margins and sentinel node biopsy, which was negative. A whole-body scan with positron emission tomography and computed tomography showed no evidence of metastatic lesions. The final staging of the patient was T4bN0M0 (Stage IIC).
Immunohistochemical examination demonstrated stronger reactivity of tumour cells to antibodies against MITF (Atlas Antibodies, Stockholm, Sweden), tyrosinase (TYR) (Cell Signaling Technology, Danvers, MA, USA), tyrosinase-related protein 1 (TRP1) (Novocastra, Newcastle upon Tyne, UK), and tyrosinase-related protein 2 (TRP2) (Abcam, Cambridge, MA, USA) than that of normal epidermal melanocytes (Fig. S1 c-h), as well as positivity to DOPA reaction (Sigma-Aldrich, Saint Louis, USA) (not shown), albeit with some heterogeneity among the tumour cells.
Genetic blood testing gave a diagnosis of OCA4, as the patient had 2 heterozygous mutations in SLC45A2 (p.P78L and p.D157N) (Fig. 2). Further testing with MM cells showed no change in mutations in SLC45A2, indicating no recovery of gene mutation in MM. Mutation of BRAF p.V600E was detected, and no NRAS (exon 2) or KIT (exon 9, 11, 13, 17, and 18) mutations were found.
Fig. 2. Mutations in SLC45A2 genes in Japanese patients with OCA. Part of this figure has been taken from Suzuki & Tomita (2). Red indicates mutations.
Postoperative adjuvant chemotherapy consisted of 3 courses of DAV-Feron immunochemotherapy (dacarbazine, nimustine hydrochloride, vincristine, interferon (INF)-β) and subsequent monthly local injection of INF-β. However, bone and lymph node metastases developed, and the patient died approximately 19 months after his initial visit.
OCA is classified into 2 types: non-syndromic and syndromic, with non-syndromic further subdivided into 7 forms based on clinical and genetic findings (1). OCA types 1 (OCA1) and 2 account for approximately 40% and 50% of OCA cases, respectively, while OCA4 is rare worldwide (2). However, OCA4 is the second-most common type following OCA1 in Far-East Asian populations, particularly among the Japanese, with a prevalence of 24–27% (2, 3). OCA4 is caused by mutations in the SLC45A2 gene (4). The SLC45A2 protein, which contains 12 putative transmembrane domains, is expressed only in melanocytes and may function as a membrane transporter in melanosomes (5). p.D157N, a mutation also detected in the present case, is the most common mutant allele in Japanese OCA4 patients, with an allele frequency of 0.39, and confers very low functional activity in melanogenesis (2). p.P78L, the other mutation, was a novel mutation. Okamura et al. (1) reported this result in 2014 and suggested that this mutation resulted in the loss of melanogenesis activity.
The majority of cutaneous malignancies in OCA are SCC (75–88%), with BCC (9–23%) being less common, while MM is rare (1.3–3%) (6, 7). To our knowledge, while 33 cases of MM in patients with OCA have been reported in the English literature, 23 cases of cutaneous malignancies with OCA were reported in the Japanese literature, of which 10 cases (43%) were MM, followed by SCC and BCC in 8 (34%) and 7 (30%) cases, respectively. As many cases may be unreported, a cohort study of OCA patients is required for the correct relative frequencies in Japan. To our knowledge, this is the first case with melanotic MM in OCA4.
Amelanotic and melanotic MM were reported at the same frequency in cases of OCA. All cases of MM with OCA presenting with the absence of tyrosinase activity, usually type 1a, were amelanotic MM. However, other types of OCA with normal or weak tyrosinase activity can develop both melanotic MM as well as amelanotic MM. In OCA4 melanocytes, tyrosinase activity is reduced due to disruption of tyrosinase processing and intracellular trafficking to the melanosome, and the enzyme is abnormally secreted from the cells in immature melanosomes (2, 8). MITF in melanocytes controls the expression of various genes that are essential for normal melanin synthesis, such as TYR, TRP1, and TRP2, and promotes differentiation of melanocytes and increases production of melanin (9). In melanoma, MITF is reported to be a critical factor in regulating proliferation, and amplification of the MITF gene is associated with poor patient survival (9). In the present case, MITF expression, as well as TYR, TRP1, and TRP2 protein expression, were all significantly increased in MM cells. We therefore suspected that MITF was induced in the oncogenic process and thereby promoted melanogenesis as a result, although the reason for this induction is unclear. Although it has been reported that some MMs show high positive staining for MITF (10), there has been no report about MITF expression in MM patients with OCA, and further studies are necessary.
In conclusion, we present here a case of melanotic MM in a patient with OCA4. We propose that increased expression of MITF might be related to the melanogenesis in MM in patients with OCA4.


 Click to show fullsize
Click to show fullsize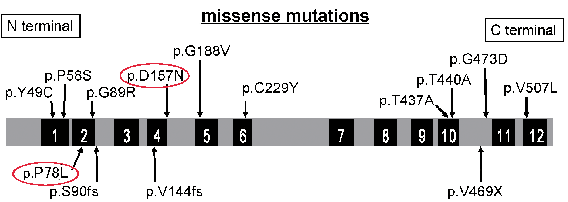 Click to show fullsize
Click to show fullsize