


Department of Dermatology, Venereology and Allergology, Wroclaw Medical University, Chalubinskiego 1, 50-368, Wroclaw, Poland. *E-mail: ola.batycka@interia.pl
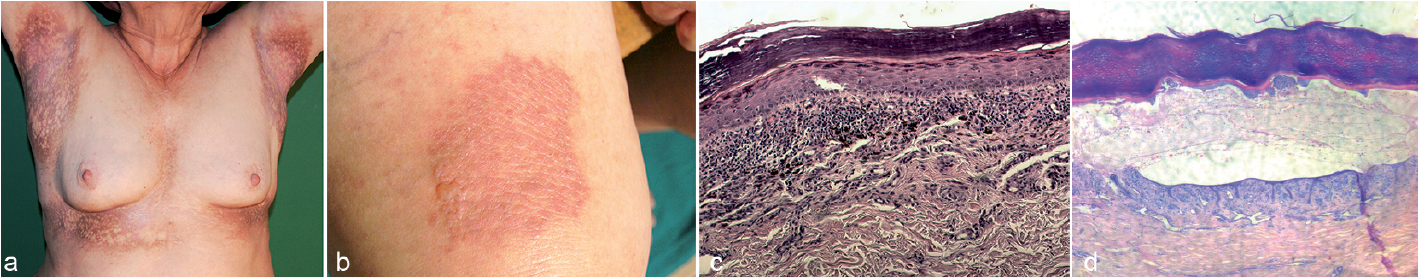
An 82-year-old woman presented with extensive, non-pruritic skin lesions, which had developed gradually over 10 years (Fig. 1a). Clinical examination also revealed shiny erythematous-violaceous plaques with several tense bullae on the forearms, thighs, dorsal aspects of the hands and feet (Fig.1b) and extensive reticular white striae on the buccal mucosa. No hair or nail abnormalities were seen. A basic laboratory work-up was normal with the exception of dyslipidaemia. Histological examinations of hyperpigmented lesions in the right groin and a bullous plaque were performed (Fig. 1c, d). Direct immunofluorescence revealed numerous cytoid bodies at the dermoepidermal junction composed of IgM, IgA and C3c in both tissue samples. No circulating anti-basement membrane zone (anti-BMZ) antibodies were found.
Fig. 1. (a) Reticular, hyperpigmented lesions in the axillae and submammary regions. (b) Tense bullae over an erythematous-violaceous plaque on the right thigh. (c) Histopathology (reticular lesion in the right groin): hyperkeratosis, hypergranulosis, vacuolar degeneration of the basal layer, band-like infiltration composed of lymphocytes and melanophages, prominent pigment incontinence (haematoxylin and eosin (H&E), original magnification ×100). (d) Histopathology (bullous plaque on the right thigh): hyperkeratosis, acanthosis, hypergranulosis, extensive bulla, band-like lymphocytic infiltration in the upper dermis (H&E, original magnification × 100).
What is your diagnosis?
Lichen planus (LP) is a chronic, inflammatory mucocutaneous disease of undetermined origin that affects approximately 1% of the population worldwide (1). In the literature numerous clinical variants of varying prevalence have been reported. Lichen planus pigmentosus (LPP) was first described by Bhutani et al. in 1974 (2). In 2001, Pock et al. (3) reported 7 patients with prominent pigmented lesions in the intertriginous areas and introduced the term lichen planus pigmentosus inversus (LPP-inversus). LPP-inversus is characterized by asymptomatic or mildly itchy hyperpigmented macules or patches with well-defined borders. The lesions are located in the intertriginous or flexural areas, including axillae, groins, inframammary folds and, less frequently, the neck and posterior auricular furrow.
The larger lesions tend to be annular or linear, and the long axis follows the lines of cleavage (3–12). Our patient presented with a rare reticular pattern of extensive hyperpigmented lesions and typical histology for LPP-inversus. This unusual clinical presentation may have resulted from the long disease duration. In cases described previously the mucous membranes as well as the scalp and nails were spared (3–12). However, in the current case the hyperpigmented lesions were accompanied by lesions in the oral mucosa.
The aetiology of LPP-inversus is unknown. No association with malignancies, hepatitis C virus (HCV) infection, medications and sun exposure has been found so far. In 2 Japanese patients described in the literature the lesions disappeared after discontinuation of wearing tight underwear, suggesting mechanical friction (Koebner phenomenon) as a triggering factor (4). However, in the case described here we could not identify any triggering factors.
Histopathologically, LPP-inversus is characterized by vacuolar degeneration of the basal layer of epidermis, slight-to-moderate lichenoid inflammatory infiltrate with prominent pigmentary incontinence in the superficial dermis, accompanied by slight or absent hyperkeratosis and hypergranulosis (3–12). Pock et al. (3) considered LPP-inversus a consequence of a rapid lichenoid reaction with intensive vacuolar degeneration of basal keratinocytes, during which compensatory proliferation of keratinocytes cannot develop. In our case we observed prominent hyperkeratosis, which may have occurred as a result of the long disease duration in an intertriginous area prone to friction (Fig.1c). Another rare clinical variant is bullous LP in which blisters develop over typical LP lesions, commonly on the lower extremities (13, 14). Histopathologically the bullae are subepidermal, but there are no linear deposits of IgG and/or C3 typical for pemphigoid.
The LPP-inversus is usually treatment-resistant; how-ever, rare cases of spontaneous resolution have been described in the literature (3, 8). Topical tacrolimus, as well as topical and oral corticosteroids have been used in most cases with limited success. Some authors recommend a “wait and watch” strategy (8, 11). In the case described here several treatment strategies were applied, including monotherapy with topical steroids, dapsone 100 mg/day or chloroquine 250 mg/day, which were found to have weak clinical efficacy. Treatment with cyclosporine A was discontinued due to hypertension. The introduction of prednisone 40 mg/day (0.5 mg/kg) in combination with dapsone 100 mg/day resulted in visible improvement and a reduction in bullae formation. However, the patient declined to continue oral steroids after 3 months of treatment due to side-effects and was lost to follow-up. To the best of our knowledge the coexistence of these 2 rare variants of LP has not been reported previously.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize