


1Service d’anatomie pathologique, 2Service d’hématologie adulte and 12Service de dermatologie, Hôpital Necker-Enfants Malade, APHP, Université Paris Descartes, Paris, 3Service d’anatomie pathologique, Hôpital Maisonneuve-Rosemont, Montréal, Canada, 4Service d’anatomie pathologique and 5Service de dermatologie, Hôpital Henri Mondor, APHP, Université Paris Est, Créteil, 6Service de dermatologie, Hôpital Cochin, APHP, Université Paris Descartes, 7UF de dermatologie, Hôpital La Pitié-Salpétrière, APHP, Université Pierre et Marie Curie, 8Service d’anatomie pathologique, 14Service de dermatologie, Hôpital Saint Louis, APHP, Université Paris Diderot, Paris, 9Service de dermatologie, CHU de Bordeaux, Université de Bordeaux, Bordeaux, 10Service d’anatomie pathologique, CHU de Bordeaux, Bordeaux, 11Registre des hémopathies malignes de Côte d’Or, Université de Bourgogne, CHU de Dijon, Dijon, 13INSERM U1163 and CNRS ERL8654, Imagine Institute, Paris, and 15INSERM U976, Hôpital Saint-Louis, APHP, Paris, France
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare condition usually considered to have a favourable prognosis. However, it is not known whether polychemotherapy or immunosuppressive-based therapy is the best approach for treating SPTCL. Using data collected between 2000 and 2012 in France, we analysed clinical, biological and pathological data of 27 patients with SPTCL. Medical history revealed that 40% of patients had been previously diagnosed with an autoimmune disorder and 22% with inflammatory panniculitis. Haemophagocytic syndrome was present in 37% of cases. Autoantibodies were positive in 65% of cases. Complete remission (CR) was reached in 74% of cases. Immunosuppressive drug treatment was given in 69.5% of patients (group 1) and polychemotherapy in 30.5% (group 2). CR was 81.2% and 28.5% (p = 0.025), respectively. Progression rate was 6.2% and 42.8% (p = 0.067), respectively. This study suggests that immunosuppressive drugs should be considered as the first-line treatment for SPTCL.
Key words: subcutaneous panniculitis-like T-cell lymphoma; chemotherapy; immunosuppresive agents.
Accepted Oct 6, 2016; Epub ahead of print Oct 10, 2016
Acta Derm Venereol 2017; 97: XX–XX.
Corr: Sylvie Fraitag, Service d’anatomie pathologique, Hôpital Necker-Enfants Malades, 149 rue de Sèvres, FR-75015 Paris, France. E-mail: sylvie.fraitag@aphp.fr
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare condition, defined as a cytotoxic T cell-mediated lymphoma of αβ-T-cell origin (SPTCL-AB) (1). Diagnostic criteria have been described in the World Health Organization (WHO) classification and γδ-T-cell lymphomas are now considered as a distinct entity (2, 3). In 2008, the European Organisation for Research and Treatment of Cancer (EORTC) conducted a major study, which highlighted the good prognosis of SPTCL and the adverse impact of haemophagocytic syndrome (HPS) on survival (4). In this study, most patients were treated with polychemotherapy, while approximately one-third received immunosuppressive drugs. However, the respective efficacy of these treatments was not evaluated. Most other cohorts have reported patients treated with polychemotherapy, where complete response (CR) was reached in 0–67% of patients (5–7). By contrast, numerous case reports or small series suggest that immunosuppressive drugs could also be effective in some cases (8–14). It should be emphasized that interpretation of data can be sometimes hampered by the possibility of misdiagnoses. Indeed, one of the main alternative diagnoses is lupus erythematosus profundus (15–17), resulting in some lupus panniculitis being misdiagnosed as SPTCL. The possible association of SPTCL with autoimmunity further increases the difficulty of diagnosis (18–21), as oligo- or monoclonal T-cell receptor rearrangement can occasionally be detected in lupus panniculitis (22). New criteria have been proposed to distinguish these 2 disorders, based on the expression of human myxovirus resistance protein 1 (MxA) (23) or the presence of plasmacytoid dendritic cell clusters (24) in lupus panniculitis, but not in SPTCL. Now that the SPTCL diagnosis criteria have been better described and clearly defined, the French Group for Study of Cutaneous Lymphoma (GFELC) favours a conservative approach, with immunosuppressive drugs as first-line treatment, as long as the patient’s health condition allows it. The GFELC has systematically reviewed all cases of SPTCL that were registered in its database according to WHO classification criteria. The aims were to better characterize the clinical and biological features of SPTCL; to review all biopsies in order to homogenize and confirm histological analysis; and to compare different therapeutic strategies in terms of response and survival. This study reports the analysis of 27 patients who were diagnosed with an SPTCL in France.
A retrospective multicentre analysis of patients with an SPTCL diagnosed between 2000 and 2012 in France was conducted. Two databases were used to recruit patients. The GFELC register is composed of all cases of cutaneous lymphoma reported by a dermatologist or expert pathologist from 1 of 39 reference centres in France. All these cases had been previously reviewed and confirmed during one or several national multidisciplinary meetings. The LYMPHOPATH network (http://www.e-cancer.fr/content/download/119823/1431492/file/LYMPHOPATH-CER-et-responsables.pdf) is a national database funded by the National Institute of Cancer (INCa) (www.e-cancer.fr). It registers all new cases of lymphoma diagnosed in France since 2011, which have been submitted for review in 1 of the 31 reference centres in France. These 2 databases were crossed to exclude duplicates. Clinical and biological data were then collected in each centre and histological materials were centralized. This work was approved by the Institutional Review Board ”Ile de France II”, Paris, France (IRB registration number: 2015-05-04).
One or more biopsy samples were collected for each case. A systematic review was carried out by an experienced haematopathologist (NB) and dermatopathologist (SF). Paraffin-embedded sections were stained with haematoxylin and eosin for histopathological analysis. The following panel of antibodies was used: CD3, CD5, CD8, CD4, CD20, CD138, CD56, CD30, Granzyme B, TIA-1, p53, CD68, CD123, CD303 (Dako, Copenhagen, Denmark), Ki67 (Immunotech, Marseille, France), Mx-A (Atlas antibodies, Bromma, Sweden), and βF1 (Santa Cruz Biotechnology, Dallas, Sweden, TCRβF1). Epstein-Barr virus-encoded RNA (EBER) in situ hybridization using EBER oligonucleotides was performed on formalin-fixed sections using a Dako hybridization kit (Dako, Copenhagen, Denmark). SPTCL was diagnosed according to WHO criteria (2): lobular panniculitis, lymphocytic adipocytic rimming, lymphocytes with nuclear irregularity, nuclear debris, fat necrosis, haemophagocytosis, septal involvement. Immunostainings were interpreted as followed: negative (less than 25% of positive lymphoid cells), low (25–50%), intermediate (51–75%) or elevated (more than 75% of positive lymphoid cells). A γδ-T-cell lymphoma was excluded either by expression of βF1 on tumour cells (n = 22) or by characterization of a monoclonal β T-cell receptor rearrangement in the skin biopsy (n = 19). βF1 staining was considered positive if at least 25% of cells were stained. Staining was not evaluable in 5 patients due to the use of Bouin as fixative. To exclude lupus panniculitis, classical histological features of lupus were used, i.e. epidermal involvement and/or peri-adnexal, perivascular lymphocytic infiltrate and mucin deposition in the dermis, lymphoid follicles, immunohistochemistry showing CD20+ B and CD138+ plasma cells, and Mx-A staining on lymphocytes (23) and/or clusters of CD123+CD303+ plasmacytoid dendritic cells (24). As an internal control, 10 cases of lupus panniculitis were reviewed to validate these criteria (data not shown). HPS was defined according to HLH-2004 criteria (25).
Responses to treatment were classified as CR, partial response (PR), stable disease (SD) or progressive disease (PD), based on the International Workshop criteria (26). Patients’ overall survival (OS) was defined as the time between the date of diagnosis and the date of death or last follow-up. The duration of CR was defined as the time between the date of CR and the date of relapse or last-follow-up. Percentages were compared using Fisher’s exact test. Prognosis variables were identified with Cox proportional hazard regression model. All statistical tests were 2-tailed, with a significance level of 0.05. Analyses were performed using Prism v6.0b (GraphPad) or with R v3.0.3.
Between 2000 and 2012, 27 patients with SPTCL were registered in France (Table SI). Most cases (22/27) were diagnosed between 2008 and 2012, after the establishment of the international consensual diagnosis criteria. In this cohort, 22 were female, with a female to male (F/M) ratio of 4.4. Median age at diagnosis was 31.1 (range 1–55) years. Interestingly, 5 cases were paediatric (less than 15 years old) and 3 of these were children aged less than 3 years old. An infectious disease (toxoplasmosis, chicken pox, and viral nasopharyngitis) preceded 3 paediatric cases. Autoimmune diseases were frequent in the medical history of adult patients. Lupus erythematous was the most frequent (n = 6), and was associated with an antiphospholipid syndrome for 2 patients. Moreover, 6 patients had been followed previously for another type of panniculitis. Among them, 4 were associated with lupus and first considered as lupus panniculitis, and 2 of them progressed to histiocytic cytophagic panniculitis (HCP) before being diagnosed with SPTCL. For these patients, the diagnosis of lupus panniculitis and SPTCL occurred 5, 7, 10 and 17 years apart. In 2 other cases, HCP was the first diagnosis with no sign of lupus. Finally, SPTCL began in per- or early post-partum for 3 young women, including one with a history of lupus.
At the time of diagnosis, the most frequent clinical presentation was multiple subcutaneous nodules of small size (between 1 and 5 cm). In a few cases, lesions could be larger and sometimes infiltrated at palpation or ulcerated. Only 2 patients had a single isolated lesion. Limbs were almost always affected and were spared in only 2 cases. No other location except the limbs was observed in 36% of patients (9/25). In addition, 72% of patients had associated B symptoms (fever, asthenia or weight loss). Even if cutaneous lesions were generally the only proven location of the disease, 24% of patients had clinical adenopathies or hepato-splenomegaly (Table SII). Positron emission tomography (PET) scan was performed in 7 patients and showed hypermetabolic fixation in cutaneous lesions and detected lombo-aortic and axillary lymph nodes involvement in one of them.
Abnormal cell count and inflammatory syndrome were observed in 87% and 65% of patients, respectively. When tested, β2-microglobulinaemia was elevated, with a mean ± SD value of 4.5 ± 2 mg/l (normal value: lower than 2.5 mg/l). HPS was suspected in 10 patients (37%) and confirmed by the combination of haemophagocytosis on bone marrow biopsy with elevated ferritin or hypertriglyceridaemia. Liver perturbations were also frequent and mainly related to the presence of HPS. The main biological abnormalities are detailed in Table SIII. The detection of auto-antibodies was a frequent feature. They were observed in 65% of cases, with a predominance of anti-nuclear (titre up to 1:160), anti-cardiolipin and anti-DNA antibodies. These antibodies were present in all 6 patients with a medical history of lupus or antiphospholipid antibody syndrome (ALPS), but also in 6 patients with no documented history of autoimmune diseases.
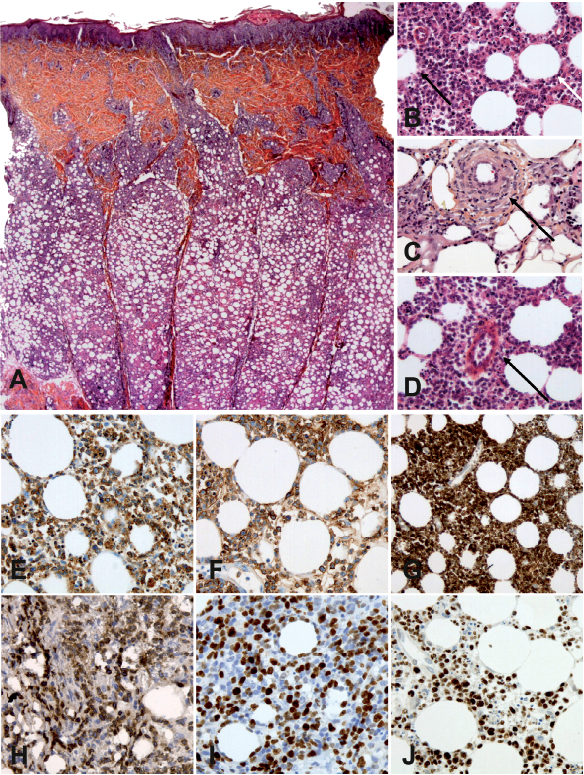
All samples exhibited a lobular panniculitis with lym-phoid cell infiltration showing irregular nuclei, adipocytic rimming, nuclear debris, lipophages and macrophages with haemophagocytosis (Fig. 1A, B). Fat necrosis was seen in 81% of patients. In 24% of cases, dermal involvement was also observed. Interestingly, vasculitis and angiotropism were noted in 4/25 (16%) and 9/25 (36%) of cases, respectively (Fig. 1C, D, respectively). In contrast to the description of lupus panniculitis, we did not observe lymphoid follicles in the lobules. Plasma cells were present in 56% of cases, mostly located in the septa.
Fig. 1. Histological and phenotypic analyses of subcutaneous panniculitis-like T-cell lymphoma (SPTCL). (A) Histological aspect (B) Subcutaneous location of SPTCL (original magnification ×100) showing atypical lymphoid cells with adipocyte rimming (white arrow) and adipocyte necrosis (dark arrow) (original magnification ×400). (C and D) Vasculitis and angiotropism of tumour cells (magnification ×400). Immunohistochemistry of SPCTL lesions (original magnification ×400) showing infiltration by cytotoxic α/β CD8 T cells expressing (E) CD3, (F) CD8, (G) granzyme B, (H) βF1, (I) Ki67, and (J) p53.
Immunohistochemistry confirmed that the infiltrates were made of cytotoxic effector CD8+ T cells expressing CD3, CD8, granzyme B and TIA1 (Fig. 1E–J). Immunophenotypical data are summarized in Table SIV. No staining was observed with CD56, CD30 or CD20, and EBV was not detected using EBER in situ hybridization. p53 staining was present in 4 patients (18%). Ki67 was elevated or intermediate in 78% of patients. βF1 staining was positive in 17/22 patients (77%) and TCR clonality was positive in 17/19 patients (89%). All evaluated patients had βF1-positive cells on biopsy, but 5 were considered negative, due to the low percentage of positive cells. All patients had at least one of these criteria and 11 patients had both criteria. For 16 patients, bone marrow biopsies were also available and only one exhibited a significant CD8+ T-cell infiltration.
Lupus panniculitis was excluded by histopathological examination with the help of MxA staining (23) and CD123/CD303 staining for plasmacytoid dendritic cells (PDC) (24). MxA staining was absent or rare in 82% of SPTCL samples (14/17), whereas 9/10 lupus panniculitis cases exhibited strong staining on lymphocytes, macrophages and PDC (p = 0.0008). In addition, no cluster of CD123+CD303+ PDC was observed in SPTCL cases (0/17), despite the presence of rare isolated PDC in 7/17 cases. By contrast, 9/10 cases of the lupus panniculitis showed clusters of PDC (p = 0.018). In combination with the absence of lymphoid follicles and B cells, these results excluded the main histological differential diagnosis of lupus panniculitis.
Median follow-up was 37.6 months after diagnosis. In this cohort, patients were mainly given first-line treatment with immunosuppressive drugs (69.5%) or with conventional polychemotherapy (30.5%). CR was obtained in 74% of patients (17/23) and the 2-year overall survival was 96% (22/23). Median follow-up after CR and CR duration was 26.6 months. In univariate analysis, the main predictive factor of CR was the use of an immunosuppressive drug as a first-line treatment (odds ratio (OR) 10.8 (95% CI 1.3–85.4), p = 0.025). Autoimmune medical past history (p = 0.15), elevated β2-microglobulin (p = 0.99), elevated lactate deshydrogenase (LDH) (p = 0.99) and the presence of HPS (p = 0.99) did not affect CR rate.
To compare these 2 treatment regimens, patients were divided into 2 groups depending on the first treatment received: immunosuppressive drugs (group 1, n = 16) or polychemotherapy (group 2, n = 7). Treatment and outcome for both groups are summarized in Table I. CR rate was 81% and 28% in group 1 and 2, respectively (n = 0.025). Patients treated with immunosuppressive drugs (group 1) received corticosteroid alone (n = 6) or a low dose of methotrexate (n = 3), cyclosporine A (n = 2) and hydroxychloroquine (n = 5). Mean duration of immunosuppressive treatment was 3.6 years (from 0.5 to 10.4 years). Three patients treated with corticosteroid alone did not reach CR, among whom 2 had SD and one achieved PR. One patient treated with hydroxychloroquine had progressive disease and was then treated with conventional polychemotherapy and reached CR. In group 2, patients received anthracycline-based chemotherapy (CHO(E)P or CHOP-related chemotherapy) and one had an autologous stem cell transplantation. Only 2 of them achieved CR. Two patients with progressive disease after polychemotherapy eventually achieved CR following treatment with corticosteroid and low-dose methotrexate. One patient died after an infectious complication and 2 continued treatment with polychemotherapy. Both groups did not differ in incidence of autoimmune medical history (p = 0.37), B symptoms (p = 0.27), β2-microglobulin (p = 0.5) or HPS incidence (p = 0.17).
Table I. Treatment and outcome of patients with subcutaneous panniculitis-like T cell lymphoma
This multicentre, retrospective, cohort study of 27 patients diagnosed with SPTCL and treated with either immunosuppressive drugs or polychemotherapy is the largest series of patients reported since the first published study of the EORTC group (4). The results show that SPTCL is a rare disease, which mainly affects young patients, especially women. This study highlights the close link between SPTCL and autoimmunity, as 40% of patients had a medical history of autoimmune disorder and 65% had autoantibodies at the time of diagnosis. A better response rate occurred with immunosuppressive drugs used as a first-line treatment rather than polychemotherapy.
SPTCL is a rare disease and few epidemiological data are available. With the collaboration of the GFELC and the LYMPHOPATH national network, we considered that we were able to collect data on almost all cases of SPTCL diagnosed in France since 2008. Sex ratio revealed an over-representation of females, with a median age of 30 years in SPTCL, a result previously reported in other series of SPTCL (4, 6, 7). This epidemiology differs from other primary cutaneous T-cell lymphoma, in whom the F/M sex ratio is close to or less than 1, and the median age is greater than 50 years (27). SPTCLs also differ from other cutaneous lymphoma by the incidence of autoimmune manifestations. In this series, almost half of the patients had an autoimmune history, which was also reported by the EORTC study, despite a lower incidence (4). The most frequent disease in our study and in the literature was lupus erythematosus (16), but many other autoimmune diseases were described in association with SPTCL, such as sarcoidosis (19), dermatomyositis (20), Sjögren’s syndrome (21), and rheumatoid arthritis (4, 28). More surprisingly, we found that 65% of patients had autoantibodies, but that half of these patients did not have a past history of autoimmune disease. Our results confirm the close link between SPTCL and autoimmunity, and suggest that the presence of autoantibodies should raise the hypothesis of an autoimmune disease with T-cell lymphoproliferation. It also raises the question of the patho-physiology of SPTCL, which might be close to those of other autoimmune diseases, as has been highlighted by microarray analysis (29). Interestingly, this study and another (30) have highlighted the potential involvement of the CCR5/CCL5 pathway in SPTCL, which could represent a therapeutic target. In children, 3 cases were diagnosed within a short period after an infectious process. Huppmann et al. (31) have highlighted the frequent association of paediatric SPTCL with autoimmune diseases or genetic/developmental abnormalities. The unusual age of these patients, the existence of a triggering factor and the short period to the beginning of SPTCL, raise the question of a clonal lymphoproliferative diathesis. An interesting hypothesis would be that SPTCL might be the consequence of a dysregulation of T cell responses after autoantigen or infectious antigen recognition. However, it is still not fully understood to which extent, or in which situation, SPTCL should be considered as a reactive T-cell lymphoproliferative disease or as a lymphoma.
As discussed in the literature, the association between SPTCL and lupus erythematous also raises a problem of misdiagnosis (32). Absence of epidermal and dermal involvement, of lymphoid follicles, of B or plasma cells, presence of cellular atypia or monoclonal TCR rearrangement are generally used to distinguish lupus panniculitis and SPTCL. However, none of these criteria are specific and can be described in overlapping form in both diseases (15, 18, 22, 32). For example, we and others (31) have reported numerous plasma cells in SPTCL, suggesting that this criterion cannot be used only for lupus diagnosis. Furthermore, the presence of a monoclonal TCR rearrangement in the skin can be observed in lupus panniculitis (22) or in other inflammatory dermatitis (19), and is not pathognomonic of T-cell lymphoma. We observed that some patients with well-documented lupus panniculitis could progress to SPTCL with monoclonal TCR rearrangement after more than 10 years of follow-up. This suggests that continuity between both entities within a spectrum of diseases might exist and demonstrates the requirement for repeated biopsies with investigation for TCR clonality throughout the follow-up. This large spectrum of diseases had been described by Magro et al. (22), and clearly highlights the challenge faced by this field for correct diagnosis. Recently, new immunohistological criteria have been described to provide help for differential diagnosis (23, 24). In lupus, MxA expression is induced on macrophages and lymphocytes in response to interferon secretion. In SPTCL, MxA is absent or is expressed on a minority of cells, mainly consisting of macrophages (23). Moreover, clusters of plasmacytoid dendritic cells have been associated with the diagnosis of lupus and were not observed in SPTCL (24). We confirmed that MxA staining and cluster of CD123+CD303+ plasmacytoid dendritic cells were rare or absent in SPTCL and can be useful tools for pathologists. Altogether, this demonstrates that diagnosis of SPTCL might be difficult and must be distinguished from γδ-T-cell lymphoma with the help of βF1 staining or monoclonal αβTCR rearrangement, but also from lupus panniculitis. These new immunophenotypical criteria could provide a significant help to pathologists to distinguish SPTCL from lupus panniculitis or γδ-cutaneous T-cell lymphoma (Fig. 2).
Fig. 2. Proposition of algorithm for subcutaneous panniculitis-like T-cell lymphoma diagnosis and treatment. PDC: plasmacytoid dendritic cells; TCR: T cell receptor; γδ-TCL: γδ-T-cell lymphoma.
SPTCLs are considered as having a good prognosis, although reported overall survival varies between 64% and 82% (4–7). The most commonly used treatment is polychemotherapy, with CR reached in 0–67% of reported cases. This variability could reflect differences in disease severity from one study to another, as well as differences in therapeutic strategies. There is no clear consensus about the best therapeutic approach, as other treatments, such as radiotherapy or immunosuppressive drugs, were also reported. Efficacy of immunosuppressive drugs in SPTCL was mainly reported in small series or case reports, as first-line (8, 9, 12) or salvage therapy (10, 33–35). Considering the increasing number of cases achieving CR described with immunosuppressive drugs, even after failure of polychemotherapy, we compared both strategies in first-line treatment. We were surprised to find that CR was reached in 81% of patients treated with immunosuppressive drug and only 28% with polychemotherapy. We are aware that this difference could be biased by the choice of physicians to treat more severe diseases with polychemotherapy. However, we did not observe any significant differences between both groups of patients in terms of clinical presentation, biological abnormalities or, importantly, incidence of HPS. HPS has been described as a major prognosis factor and has been associated with a lower 5-year overall survival (4). In our study, due to low mortality rate, we failed to identify HPS as a prognosis factor for survival. Interestingly, HPS had no impact on CR rate. This difference could be explained by the size of our series, which could be insufficient to demonstrate the impact of HPS. However, more patients were treated with immunosuppressive drugs in this study than in the EORTC’s one, and the efficiency of this treatment in SPTCL may have minimized the influence of HPS on response rate. It is of importance to note that we identify a higher proportion of patients with HPS in our cohort, by comparison with EORTC’s data (37% vs. 17%). Again, this could be biased by the size of our cohort. However, other studies also reported high frequency of HPS in SPTCL (36). To avoid any misevaluation of patients, we considered that patients had HPS only when they reached HLH-2004 criteria (25). Interestingly, the 2 patients who had progressive diseases after polychemotherapy and reached CR with immunosuppressive drugs in salvage therapy also had HPS. The most frequent immunosuppressive drugs were corticosteroids in combination with methotrexate or cyclosporine A. Stable or progressive disease were only observed with hydroxychloroquine or corticosteroids alone. Even if there is no other large study evaluating the superiority of one of these treatments, our series and literature analysis suggest that corticosteroids in combination with methotrexate or cyclosporine A might represent a good strategy in first-line therapy for SPTCL.
Altogether, this study strongly suggests that immunosuppressive drugs should be considered as first-line therapy for patients with SPTCL, even if some of them could still benefit from polychemotherapy in case of treatment failure. Especially in the cases of patients with HPS, polychemotherapy should be considered as an alternative in the absence of rapid response to immunosuppressive drugs. Moreover, efficacy of immunosuppressive drugs in this pathology, and the frequency of autoimmune biological abnormalities or the occurrence of infections before their onset in children, clearly raised the question of the physiopathology of SPTL. Further studies could help to determine if SPTCL is a lymphoma induced by chronic antigen stimulation in autoimmune diseases, or if it may be rather considered as a reactive T-cell lymphoproliferative syndrome than a true T-cell lymphoma.
The authors would like to thank all members of the French Group for Study of Cutaneous Lymphoma (GFELC) and the LYMPHOPATH network for their participation in this study. The authors also wish to thank Corinne Haioun, Guy Leverger, Juliette Mazereeuw-Hautier and Yves Perel for their help during data collection and for insightful discussions. The authors would like to thank the association ”Des tulipes contre le cancer”.
All of the following authors contributed to this study by providing 1 or 2 cases: Agnes Carlotti1, Camille Frances2, Nicolas Meyer3, Laurence Lamant4, Eric Frouin5, Luc Xerri6, Brigitte Balme7, Stéphane Dalle8, Julie Bruneau9,10, Olivia Boccara11. (1Service d’anatomie pathologique, Hôpital Cochin, APHP, Université Paris Descartes, 2Service de dermatologie, Hôpital Tenon, APHP, Université Pierre et Marie Curie Paris VI, Paris, 3Service de dermatologie, Institut Universitaire du Cancer, Univeristé Paul Sabatier-Toulouse III, 4Service d’anatomie pathologique, Institut Universitaire du Cancer de Toulouse, Oncopole, Toulouse, 5Service d’anatomie pathologique, CHU de Montpellier, Montpellier, 6Service d’anatomie pathologique, Institut Paoli Calmettes, Marseille, 7Service d’anatomie pathologique, CHU de Lyon, 8Institut de Cancérologie de Lyon, Centre de recherche en Cancérologie de Lyon, Lyon, 9Service d’anatomie pathologique, and 11Service de dermatologie, Hôpital Necker-Enfants Malades, APHP, Université Paris Descartes, Sorbonne Paris Cité, 10INSERM U1163 and CNRS ERL8654, Imagine Institute, Paris, France.)
The authors declare no conflicts of interests.


 Click to show fullsize
Click to show fullsize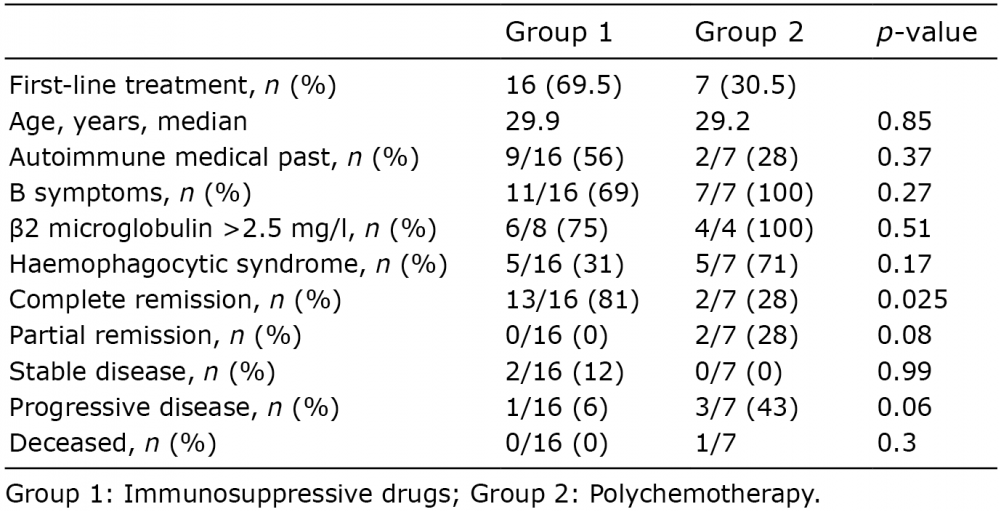 Click to show fullsize
Click to show fullsize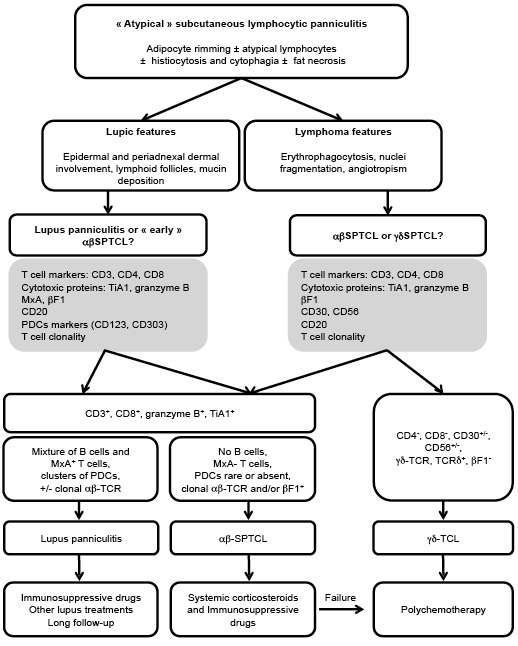 Click to show fullsize
Click to show fullsize