


Departments of 1Dermatology and 3Pediatrics, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, 2Center for Advanced Medicine and Clinical Research, Nagoya University Hospital, Departments of Dermatology, 4Nagoya City University, Nagoya, and 5Fujita Health University School of Medicine, Toyoake, Japan. *E-mail: makiyama@med.nagoya-u.ac.jp
The arachidonate lipoxygenase 3 (ALOXE3) gene is a causative gene of lamellar ichthyosis, congenital ichthyosiform erythroderma (CIE) and pleomorphic ichthyosis (PI, also called self-healing/self-improving collodion baby) (1, 2). Oxygenation of the linoleate moiety of ceramides catalysed by ALOXE3 constitutes an indispensable step in the covalent linkage of ω-hydroxyl ceramides to proteins of the cornified cell envelope and, subsequently, for the formation of the corneocyte lipid envelope. To date, 18 pathogenic mutations in ALOXE3 have been reported in autosomal recessive congenital ichthyoses (ARCI) (www.hgmd.cf.ac.uk, as of Human Gene Mutation Database professional 2016.6.27). They comprise 5 missense, 5 nonsense, 4 splice-site, 3 small deletion and 1 gross deletion mutations (Table SI) (1, 3–11). Recently, Hellström Pigg et al. (11) reported that 9 out of 132 (6.8%) ARCI patients in Scandinavia had causative mutations in ALOXE3.
All forms of ARCI are associated with significantly impaired skin barrier function, mostly due to the inability of mutated keratinocytes to produce and/or secrete the lipids required for formation of the corneocyte lipid envelope and the extracellular lipid layers in the stratum corneum. Impaired function of ALOXE3 can cause significantly defective skin barrier formation. Clinically, Wang et al. (9) reported that decreased skin barrier function in CIE caused by ALOXE3 mutation leads to cutaneous fungal infections. We describe here a previously unreported mutation in ALOXE3, p.Leu436Pro, in a Japanese female with CIE complicated with recurrent eczema and depression.
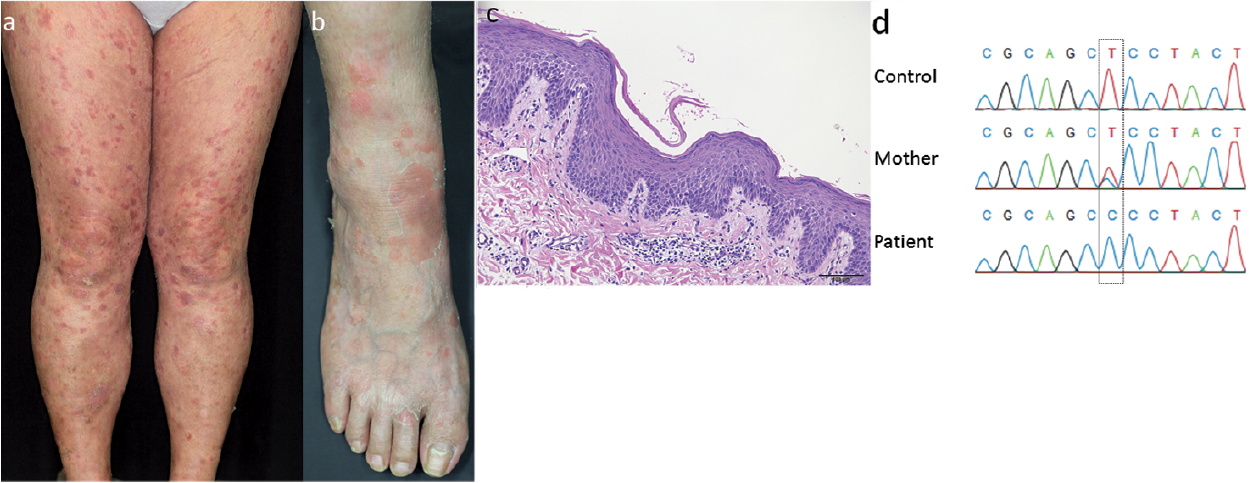
The proband is a 38-year-old Japanese woman, the first child born to first-cousin parents. She presented with generalized ery-thema and ichthyosis from one week after birth. She often showed round, scaly, itchy erythema on the trunk and extremities. Physical examination revealed multiple erythemas with fine whitish scales on the extremities (Fig. 1 a and b). Recurrent fungal infections accompanied by slight pain were seen in her skin and nails. She had been diagnosed with depression and had been treated with oral duloxetine hydrochloride, etizolam and lithium carbonate. A skin biopsy specimen from the right forearm revealed mild acanthosis, mild hyperkeratosis and slight spongiosis in the epidermis (Fig. 1c). Mild lymphocytic infiltration was seen around the vessels in the upper dermis, suggesting overlapping of mild ichthyosis and chronic eczema. She was treated with a heparinoid-containing moisturizer and an oral anti-histamine, and the eruptions improved gradually.
Fig. 1. Clinical and histo-pathological features of the proband with congenital ichthyosiform erythroderma. (a, b) Multiple round erythemas with slight pigmentation and fine whitish scaling are seen on (a) the legs and (b) the right ankle. (c) Haematoxylin-eosin staining shows partial parakeratosis, moderate hyper-keratosis and mild spongiosis in the epidermis. Dermal lymphocytic infiltration is observed. Scale bar: 100 μm (original magnification × 200). (d) Sanger sequencing reveals a homozygous mutation of ALOXE3, c.1307T>C (p.Leu436Pro), GenBank NM_021628.2, in the affected patient, and a heterozygous change in her mother.
Ethical approval was obtained and all research was performed in accordance with the principles of the Declaration of Helsinki. Genomic DNA from the patient’s peripheral blood leukocytes was used for whole-exome sequencing analysis, as described previously (12). Analysis of whole-exome sequencing data revealed the previously unreported homozygous missense mutation c.1307T>C (p.Leu436Pro) in ALOXE3, which was confirmed by Sanger sequencing (Fig. 1d). Her mother was shown to be a heterozygous carrier of this mutation (Fig. 1d). Paternal DNA was not available for analysis because the father had already died. The mutation has not been described in the Human Genetic Variation Database, which includes 1,208 exome-sequencing data sets of healthy Japanese controls, nor in the ExAC database, which includes 60,706 exome data (http://exac.broadinstitute.org/ as of 29/11/2015). T at nucleotide position 1,307 is the second base in exon 11 of ALOXE3. In silico analyses using 2 splice-site prediction tools, SplicePort (http://spliceport.cbcb.umd.edu/SplicingAnalyser.html) and Alternative Splice Site Predictor (http://wangcomputing.com/assp/), were conducted to predict whether this mutation would lead to aberrant or normal splicing; the findings suggested that this mutation results in normal splicing (data not shown). p.Leu436Pro in ALOXE3 is located in the C-terminal catalytic lipoxygenase domain, close to the pathogenic p.Leu427Pro mutation that was previously reported in a case of PI (5, 8). In silico analysis with Sorting Intolerant From Tolerant (SIFT) and MutationTaster predicts it to be “damaging”. Thus, the mutation is thought to be functionally relevant. In addition, the whole-exome data showed no putative mutations in FLG and it showed no putative mutations in the other genes previously implicated in the molecular pathology of ichthyosis.
Not only did the proband have congenital ichthyosis due to ALOXE3 mutation, but she also had depression. We previously reported a case of Darier’s disease complicated with schizophrenia (13). Darier’s disease is a rare autosomal-dominantly inherited skin disorder caused by a heterozygous mutation in ATP2A2, which is expressed in both the skin and the brain. Darier’s disease some-times shows neuropsychiatric manifestations, which are considered to be associated with ATP2A2 expression in the brain. However, ALOXE3 is predominantly synthesized in the epidermis. To date, no reports have mentioned psychiatric disorders in ARCI cases associated with ALOXE3 mutations. Recently, the burdens of inherited ichthyoses were described based on a French national survey (14). The negative impacts of ichthyoses were obvious in domestic life, educational/professional life and leisure/sports activities. Such stressors might have caused her depression.
Filaggrin, found to be normal in the reported patient, is a key protein that facilitates the terminal differentiation of the epidermis and the formation of the protective stratum corneum barrier (15). Mutations in FLG encoding profilaggrin/filaggrin are a genetic cause of ichthyosis vulgaris and are also known as a major predisposing factor for atopic dermatitis (AD). Skin barrier dysfunction and accelerated percutaneous sensitization to multiple allergens are thought to play important roles in the pathogenesis of AD in patients with FLG mutations. In addition, most patients with Netherton syndrome (NS; MIM_256500) display AD-like lesions. NS is autosomal recessive disorder caused by loss-of-function mutations in the serine protease inhibitor of the Kazal-type 5 (SPINK5) gene. Overexpression of serine proteases in a mouse model of NS leads to stratum corneum detachment, epidermal barrier dysfunction, and accelerated inflammation. Notably, the present CIE patient with skin barrier defects due to the ALOXE3 mutations also showed recurrent eczema with an elevated serum IgE level (349 IU/ml; normal range: <233 IU/ml). Thus, patients with ARCI caused by ALOXE3 mutations might have recurrent dermatitis resulting from the reduced barrier function and increased risk of percutaneous sensitization to allergens, similar to AD or AD-like lesions in patients with filaggrin deficiency and in patients with NS. Furthermore, this case had recurrent episodes of cutaneous fungal infection, as was seen in a previously reported patient with ARCI (9).
This study was supported by JSPS KAKENHI Grants Numbers 15H04887, 15K15415, 15H06280, 16K19717. The work was also supported by the Japan Intractable Diseases Research Foundation. The authors acknowledge the Human Genome Center, Institute of Medical Science, the University of Tokyo (http://sc.hgc.jp/shirokane.html) for providing super-computing resources.


 Click to show fullsize
Click to show fullsize