


1Department of Dermatology, Hyogo College of Medicine, 1-1, Mukogawa-cho, Nishinomiya, Hyogo, 663-8501, 2Division of Dermatology, Department of Internal Related, Kobe University Graduate School of Medicine, Kobe, and 3Department of Dermatology, Hyogo Prefectural Kaibara Hospital, Tanba, Hyogo, Japan. E-mail: kyamanis@hyo-med.ac.jp
Accepted Oct 12, 2016; Epub ahead of print Oct 14, 2016
Autosomal recessive congenital ichthyosis (ARCI) is an entity of rare genetic skin disorders with abnormal keratinization (1). Harlequin ichthyosis is the most severe type of ARCI, usually caused by truncation mutations in ABCA12. In subtypes of ARCI, such as lamellar ichthy-osis (LI) and, congenital ichthyosiform erythroderma (CIE) mutations in TGM1, ABCA12, NIPAL4, ALOXE3, ALOX12B, CERS3, CYP4F22 and PNPLA1 have been reported (2–4). Mutations in NIPAL4, the gene encoding the NIPA-like domain containing 4 protein (NIPAL4/ichthyin) (5), have been identified in approximately 16% of patients with ARCI (2). We report here a case of ARCI with variable CIE symptoms. This is the first reported case with a mutation in NIPAL4 in East Asia.
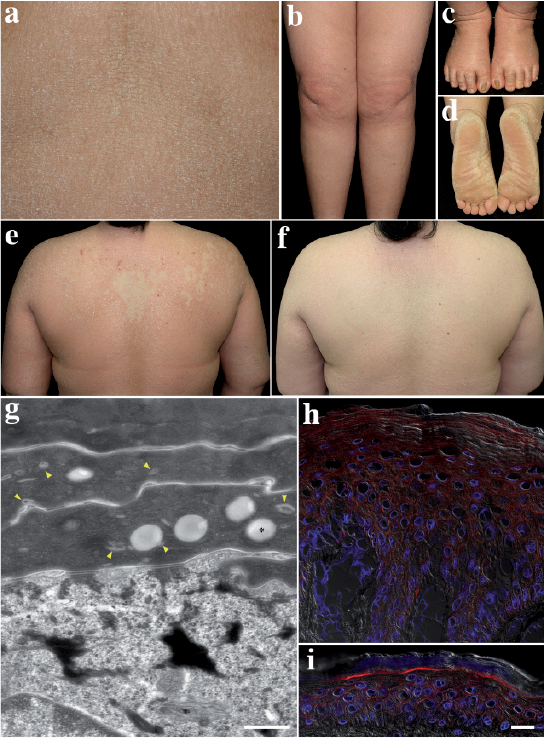
A 41-year-old Japanese woman had had generalized dry skin and desquamation since birth. Her younger sister also had a similar skin condition with atopic dermatitis (AD), but the complication of AD was absent in the present case. No collodion baby phenotype was reported at birth in either sister. No other members of the patient’s family had a history or symptoms of ichthyosis. On examination, the patient had erythroderma with generalized fine scales and occasional itching (Fig. 1a, b). She showed mild ectropion. Elabium was absent. The nails of both her first toes showed onychogryphosis (Fig. 1c), and her fingernails were mildly thickened with transverse ridges. Palmar hyperkeratosis was mild, but her soles were diffusely covered with thick scales (Fig. 1d). She reported anhidrosis and heat exhaustion in the summer months. Interestingly, a mild geographic lesion was distributed from her right shoulder to the centre of the upper back at her first visit (Fig. 1e). The lesions were surrounded by infiltrated diffuse ery-themas with coarse scales and several small erosions, possibly due to scratching, were noted. However, 6 months later, the erythroderma had improved and milder ichthyosiform lesions were diffusely distributed over her entire body surface (Fig. 1f). Such variations in her erythroderma and scales were repeated and were not necessarily dependent on the season or sweating. Histology of a biopsy from her left upper arm showed epidermal acanthosis and a homogeneously thickened stratum corneum (Fig. S1a). Loss or hypoplasia of granular layers and granular degeneration were absent. Several perivascular inflammatory infiltrates were noted. As differential diagnoses, ichthyosis vulgaris and epidermolytic ichthyosis were excluded, based on the histology, and congenital ichthyosiform erythroderma was diagnosed.
Fig. 1. Clinical, ultrastructural and immunoflourescence findings. Mild ichthyosiform erythroderma with fine scales distributed on: (a) the abdomen; and (b) the legs. (c) Thickened and deformed nails of both first toes. (d) Diffusely hyperkeratotic soles with coarse scales. (e) Milder lesions surrounded by infiltrated diffuse erythemas with coarse scales. (f) Six months later, the skin condition was milder. (g) Ultrastructure of the epidermis. Lipid droplets (asterisk) and several small membranous structures (yellow arrow-heads) are visible in the stratum corneum. Bar=0.5 μm. (h, i) Immunofluorescence for GluCer in: (h) healthy control skin, and (i) the patient’s skin. A rabbit anti-GluCer antibody (1:50 dilution) (Glycobiotech, Borstel, Germany) and reagents shown in the Fig. S1 legend were used. Images are merged views of GluCer (red), DAPI (blue) and differential interference contrast (DIC) images (white). Bar = 20 µm.
Studies using human tissues and/or DNA were performed with approval from the ethics committee of the Hyogo College of Medicine (permit numbers: hi32 and 212) and written informed consent was obtained from the patient and all donors. Immunofluorescence analysis using a NIPAL4/ichthyin antibody raised against a C-terminal portion of the protein was positive in the upper spinous and granular layers of normal epidermis (Fig. S1b), and that staining pattern was similar in the acanthotic epidermis of the patient (Fig. S1c). Ultrastructural analysis revealed lipid droplets and several small membranous structures, possibly remnants of lamellar granules, in the stratum corneum (Fig. 1g), but vesicular structures and abnormal lamellar granules, which were reported previously (6), were not clearly evident. Glucosylceramide (GluCer) was faintly, but diffusely, stained in the epidermis including the stratum corneum of the lesion (Fig. 1h), in contrast with the intense staining of GluCer beneath the stratum corneum of healthy skin (Fig. 1i). Whole-exome sequencing of the patient’s genomic DNA revealed a novel homozygous sequence variation, which corresponds to c.458G>A (NM_001099287) in the NIPAL4 gene (Fig. S2a). That sequence variation was not reported in the Integrative Japanese Genome Variation Database (iJGVD) or in dbSNP (release Build 147). No mutations were found in any of the other genes responsible for ARCI. Sanger sequencing revealed that the patient and her sister had the same homozygous mutation of c.458G>A, while her mother and her 2 children were heterozygous for the mutation and her husband was homozygous for c.458G= (Fig. S2b). Thus, the mutation was completely linked to the phenotype of the disease. The patient was mainly treated with topical emollients and petrolatum.
In the present Japanese case with CIE, we identified a novel homozygous mutation c.458G>A in the NIPAL4 gene that leads to a p.Arg153Gln mutation in the NIPAL4/ichthyin protein. As summarized in Fig. S3, the NIPAL4 mutations reported in ARCI patients are distributed over the coding sequence and/or splice sites. The mutation c.458G>A is in exon 2 as are c.403A>C, c.425G>T and c.433C>T, while p.Arg153Gln is in the first cytoplasmic loop of the protein near p.Gly142Val or p.Arg145*. ARCI patients with NIPAL4 mutations have or CIE with or without a collodion membrane at birth, ectropion or palmoplantar keratoderma, which vary unexpectedly even among patients with the same homozygous mutation of c.527C>A (7). The present case was characterized by a variable manifestation of ichthyosiform erythroderma with fine scales, which become severe or less severe independently of the season. The present case had no characteristic signs of AD, but the affected sister was associated with AD. Wajid et al. (8) reported that some members with the NIPAL4 p.Ala176Asp mutation develop AD in Pakistani families.
NIPAL4/Ichthyin is thought to function as a Mg2+ transporter or to play a role in lipid processing in the epidermis, although its precise function is unknown (5). In our case, staining for NIPAL4/ichthyin was clearly positive in the epidermis of the patient’s skin as well as in healthy skin. Hence, the mutation of the present case may not affect the stability of that protein. Different patterns of staining, at the cell periphery in healthy skin or both at the cell periphery and in the cytoplasm in NIPAL4-mutated skins (9, 10), were not necessarily evident in the present case. In contrast, the stratum corneum in the patient’s skin was positive for GluCer, but not in healthy skin, where it is degraded to ceramides to form intercellular lipid lamellar structures. The remaining GluCer immunoreactivity in the patient’s stratum corneum seems to coincide with the accumulation of lipid droplets seen at the ultrastructural level and suggests a defective extracellular release and processing of GluCer in epidermis.
The authors thank members of the Joint-Use Research Facilities of the Hyogo College of Medicine for their technical assistance. This work was supported in-part by JSPS KAKENHI Grant Number 26461668.


 Click to show fullsize
Click to show fullsize