


Departments of 1Dermatology and 4Pediatrics, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan, 2UCL Medical School, University College London, UK, 3Center for Advanced Medicine and Clinical Research, Nagoya University Hospital, Nagoya, 5Department of Dermatology, Fujita Health University School of Medicine, Toyoake, and 6Department of Dermatology, Juntendo University Urayasu Hospital, Urayasu, Japan. *E-mail: makiyama@med.nagoya-u.ac.jp
Accepted Oct 26, 2016; Epub ahead of print Oct 27, 2016
Palmoplantar keratoderma (PPK) is a group of inherited skin disorders characterized by thickening of the skin on the palms and soles, often leading to pain. Classification has largely been clinical, but recently, causative genes for the various PPKs have been found, resulting in a need for a genetic classification of PPK (1). The most common type in the Japanese population is Nagashima-type PPK (NPPK) (2). This autosomal recessive condition is caused by a mutation in SERPINB7. The SERPINB7 protein is found in the stratum corneum of the skin, and its main function is to regulate intercellular protease activity (3). The clinical features of NPPK include diffuse hyperkeratosis associated with erythema on the palms and soles and, in addition, on the dorsal aspects, known as transgrediens (2). Another type of PPK, striate PPK (SPPK), is autosomal dominant. The clinical features include strips of hyperkeratosis running longitudinally along the hands and fingers, and generalized hyperkeratosis on the soles of the feet. A mutation in any of 3 genes is reported to be an underlying cause: desmoglein 1 (DSG1), desmoplakin (DSP) and keratin 1 (KRT1) (4). Not only are these desmosomal proteins essential in maintaining the structure and integrity of the skin, but they are also essential to cell turnover (5). DSP mutations have been shown to increase skin proliferation, leading to hyperkeratosis, and the same effect has been predicted with DSG1 mutations (1), although the exact mechanism has not been fully clarified despite DSG1 being involved in various skin disorders (6, 7). To date, there have been no reports of a patient with genetic mutations for 2 types of PPK. Here, we describe such a Japanese male.
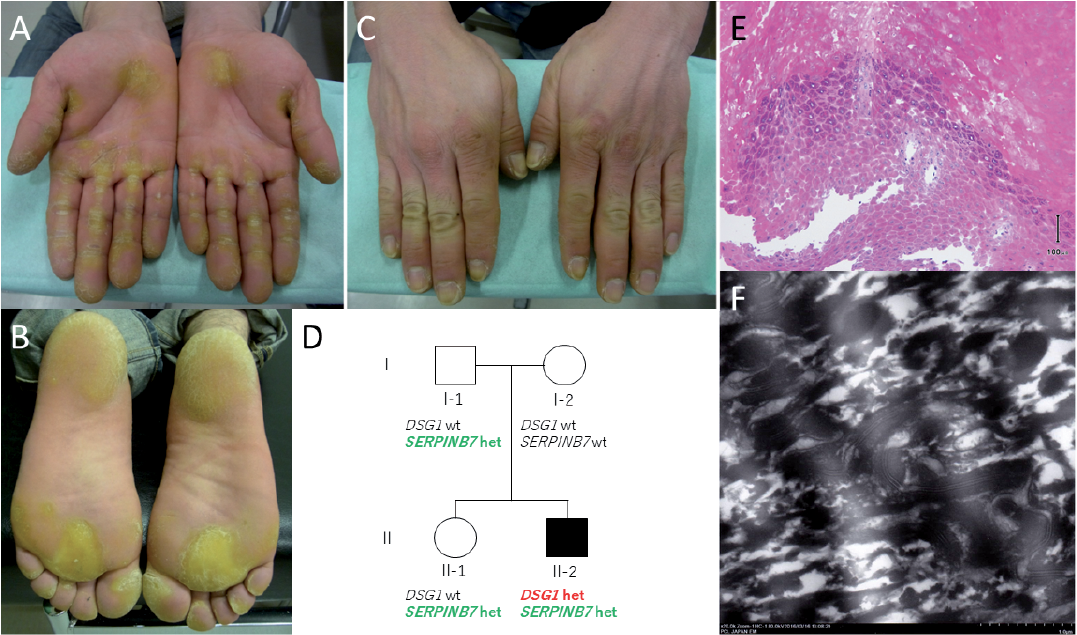
A 36-year-old man presented with longstanding pain and hyperkeratotic palms. He had presented with corns on his palms in adolescence, which developed easily after sports. It was when he started working as a farmer that he noticed pain and “stripe-like” hyperkeratosis on his palms, and circular areas of hyperkeratosis on the soles (Fig. 1A and B). He also had abnormalities on the dorsal aspects of the hands: poorly demarcated areas of erythema and maceration (Fig. 1C). No lesions were found on the dorsa of the feet. Although there were no white spongy changes after water immersion, as has been reported for some cases of NPPK, his palms were abnormally moist and malodorous compared with a healthy subject. The stripe-like pattern of hyperkeratosis on this patient’s palms appeared to be a classical example of SPPK. However, the continuation of the lesions to the dorsal aspects, and a change in the lesions from hyperkeratosis to erythema were atypical. Instead, these seemed to resemble transgrediens reported in NPPK (2). Other than a sister who has atopic dermatitis, he has no family history of hyperkeratosis or other skin disorders.
Fig. 1. Clinical and histological features of the patient. (A) Palms showing typical longitudinal hyperkeratotic lesions on the fingers. (B) Plantar aspects showing diffuse areas of hyper-keratosis. (C) Dorsal aspects of the hands, showing generalized erythema. (D) Pedigree of the family showing genotypes for the mutations in DSG1 and SERPINB7. wt: wild type, het: heterozygote. (E) Skin biopsy sample from the sole shows keratohyalin granules of various sizes, and acantholysis. Scale bar: 100 µm. (F) Electron microscopy clearly shows a highly electron-dense line. Magnification: ×20,000.
Informed consent and ethical approval were obtained, and all research conformed to the principles of the Declaration of Helsinki. Given the wide range of differentials and the variety of genes these conditions cover, we opted to perform whole-exome sequencing (WES) to determine the causative gene. This showed that not only did the patient have a heterozygous mutation c.655C>T (p.R219*) in the DSG1 (DSG1: NM_001942), but he was also heterozygous for a mutation c.796C>T (p.R266*) on the SERPINB7 (SERPINB7: NM_001040147) (3). The WES data excluded pathogenic mutations in other PPK-causative genes, including KRT1 and DSP. His father and sister were carriers for the SERPINB7 variant, while his mother showed a wild-type genotype (Fig. 1D). All relatives demonstrated a wild-type DSG1 variant.
Histologically, parts of the basal layer and the stratum spinosum are missing; however, we observed size irregularities in the keratohyalin granules, and acantholysis, possibly due to reduced epithelial intercellular cohesion, which is thought to result from the DSG1 mutation (Fig. 1E) (1, 8). Electron microscopy revealed, within the hyperkeratotic regions, thickening of the desmosomal intermediate zone, giving the appearance of a highly electron-dense line (Fig. 1F). Clinically, we initially suspected KRT1 mutation-induced SPPK with a V2 frameshift, which has been reported to cause SPPK (9). Histologically, it was necessary to consider a DSG1 mutation, as there have been reports of this mutation leading to acantholysis (10).
It is not clear in the presented case if transgrediens appeared because the SERPINB7 mutation directly caused the phenotype, or whether the SERPINB7 mutation had a modifying effect on DSG1 that coincidentally manifested as transgrediens on this occasion. In any case, our results suggest that simultaneous disruption of the skin by 2 different genes may create an overlap in symptoms of their respective disorders. Thus, there is a possibility that a heterozygous SERPINB7 mutation may be considered as a modifying factor for patients with DSG1 mutation-positive SPPK, although we cannot exclude the possibility that the SERPINB7 mutation had no effect on the phenotype in the heterozygous state. Indeed, the present SERPINB7 mutation c.796C>T (p.R266*) might be entirely coincidental given the high prevalence of this particular mutation in the Japanese population (see below). In addition, the protein string interactions suggest a possible link between DSG1 and SERPINA4 (one of the Serpin family members), but not SERPINB7 (11).
Another noteworthy point is that the 2 mutations were found by chance. Had we taken the usual protocol and tested only for DSG1 by Sanger sequencing on the suspicion of SPPK from the patient’s symptoms, the result would have been positive. It is likely then that we would have terminated any further investigations and initiated management for standard SPPK. On the other hand, had we only tested for SERPINB7, based on transgrediens demonstrated by the patient, the result would have shown heterozygosity. Although heterozygous SERPINB7 mutation carriers have not been reported to be symptomatic of NPPK, it is probable we would have assumed that the symptoms were due to another related mutation that was difficult to pick up by Sanger sequencing, and we would have searched in vain for a large deletion, an intronic mutation or a promoter site mutation. It was only because we opted to perform WES that we were able to visualize both mutations. We feel this may provide another angle from which to consider the genetic aetiology of PPKs. Undetected, unsearched-for modifying mutations in other PPK-causative genes, such as SERPINB7 in the present case, may be present in some previously reported, genetically defined, PPK cases. Indeed, the carrier rate of the present SERPINB7 mutant allele, c.796C>T, is estimated as 0.9% from both our screening data in the Japanese population (data not shown) and HGVD (Human Genome Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html).
In summary, we reported a patient presenting with autosomal dominant SPPK caused by the DSG1 mutation and a previously reported SERPINB7 mutation, the causative gene of autosomal recessive NPPK.
This study was supported by JSPS KAKENHI Grants #15H04887, #15K15415, #15H06280 and #16K19717. The work was also supported by the Japan Intractable Diseases Research Foundation. The authors acknowledge the Human Genome Center, Institute of Medical Science, University of Tokyo (http://sc.hgc.jp/shirokane.html) for providing supercomputing resources.


 Click to show fullsize
Click to show fullsize