


1Department of Dermatology, 2Institute of Pathology, and 3Rheumatology Unit, Rabin Medical Center – Beilinson Hospital, 4941492 Petach Tikva, and 4Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel. E-mail: shanyshnush@gmail.com
Accepted Nov 8, 2016; Epub ahead of print Nov 10, 2016
Eosinophilic granulomatosis with polyangiitis (EGPA), also known as Churg-Strauss syndrome, is a systemic vasculitis affecting small-to-medium-size vessels. It is characterized by asthma, followed by hypereosinophilia and, in the final phase, necrotizing vasculitis with extravascular granulomas and tissue infiltration by
eosinophils. Since anti-myeloperoxidase anti-neutrophil cytoplasmic antibodies (ANCA) are present in 30–40% of patients, EGPA is classified as an ANCA-associated vasculitis (1, 2).
Cutaneous lesions are a prominent feature of the vasculitic phase of EGPA, occurring in approximately 60% of patients (3). Palpable purpura is the predominant cutaneous manifestation. Others include infiltrated plaques, erythematous papules, cutaneous or subcutaneous nodules, urticaria, livedo reticularis, and digital gangrene (4).
Reports of EGPA presenting as vesicles or bullae are rare and their histological characteristics and correlation between clinical, serology and histopathology findings are scarcely described (3–6). We describe here a patient with bullous manifestations of relapsed EGPA and the corresponding histopathological findings.
A 47-year-old man was referred by his rheumatologist to the dermatology department for evaluation. The patient described a pruritic rash that had appeared one week previously over his inner arms, which had spread rapidly to his wrists and legs. He also complained of mild dyspnoea. Past medical history was remarkable for ANCA-negative EGPA, diagnosed 2 years earlier, which manifested as perimyocarditis, recurrent asthma exacerbations, mononeuritis multiplex, recalcitrant diarrhoea, palpable purpura on the shins, and elevated blood eosinophil count of up to up to 6 × 103/μl. There were histopathological findings of intestinal eosinophilic infiltrates. The patient had initially been treated with systemic steroids and cyclophosphamide, followed by courses of azathioprine and biannual rituximab.
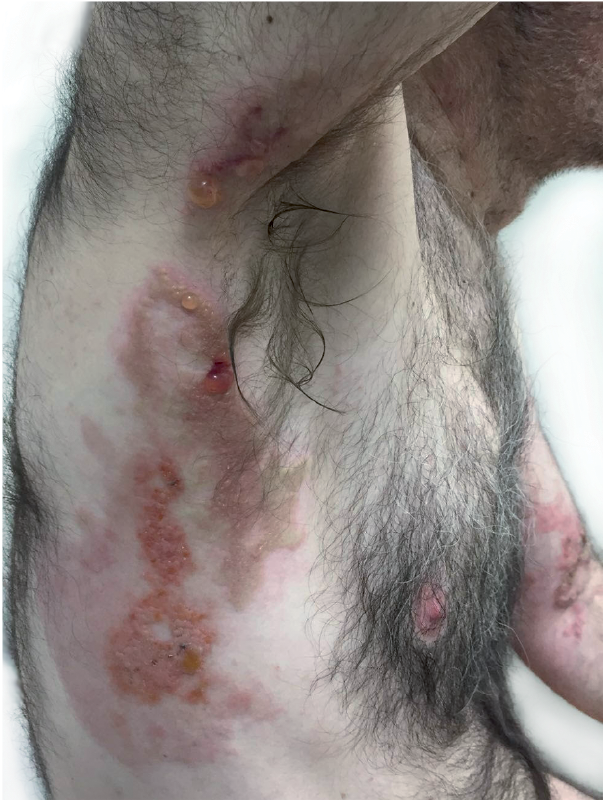
On admission, the patient was afebrile. Physical examination revealed expiratory wheezes over both lung fields and a symmetrical blistering figurate eruption involving the axillae, inner arms, wrists, and lower limbs. The rash consisted of indurated and oedematous erythematous-to-orange plaques overlaid by grouped, clear-fluid, tense bullae and vesicles in a linear and annular arrangement at the periphery of the lesions (Fig. 1). Blood tests revealed an eosinophil count of 4.9 × 103/μl. Repeated cultures and polymerase chain reaction tests from blister fluid were negative for bacterial and herpetic infections. Perilesional direct immunofluorescence showed no deposition of antibodies. Biopsy from a representative bullous lesion revealed marked oedema in the papillary dermis leading to a subepidermal blister (Fig. 2A).
Fig. 1. Eosinophilic granulomatosis with polyangiitis and a bullous rash. Note the vesicles overlying the oedematous plaques.
Fig. 2. Biopsy from a representative bullous lesion. (A) Marked oedema in the papillary dermis with subepidermal blister. Throughout the dermis there are foci of eosinophilic collagen fibres and eosinophilic deposits (i.e. “flame figures”) along mixed inflammatory cell infiltrates containing many eosinophils (Haematoxylin and eosin (H&E) × 100). (B) Higher magnification showing foci of eosinophilic collagen fibres and eosinophilic deposits, along numerous eosinophils, neutrophils and nuclear dust (H&E × 200). (C) The eosinophilic collagen fibres and eosinophilic deposits with eosinophils, neutrophils and nuclear dust, are surrounded peripherally by histiocytes, including a multinucleated giant cell (arrow) (H&E × 400). (D) A non-necrotizing granuloma in the dermis composed of epithelioid histiocytes, multinucleated giant cells and eosinophils (H&E × 400).
Throughout the dermis there were foci of eosinophilic collagen fibres with eosinophilic deposits (i.e. “flame figures”), along with many eosinophils, few neutrophils and nuclear dust (Fig. 2B, 2C). At the periphery of these foci, there were variable numbers of histiocytes and occasional multinucleated giant cells (Fig. 2C). There was also a non-necrotizing granuloma composed of epithelioid histiocytes, multinucleated giant cells and eosinophils (Fig. 2D). A diagnosis of cutaneous EGPA was made and prednisone treatment was increased from a maintenance dosage of 20 mg to 40 mg for 10 days, followed by a gradual tapering down, leading to rapid clearance of the lesions.
Two months later, the patient was hospitalized due to perimyocarditis with pericardial tamponade. Pericardial biopsy showed granulomatous infiltrates and flame figures similar to the findings in the skin biopsies. After an additional 2 months of follow-up, the patient is stable and there is no recurrence of the rash.
The characteristic histopathology of cutaneous manifestations of EGPA (3–6) includes dermal leukocytoclastic or eosinophilic vasculitis, eosinophilic infiltrate, and granuloma formation. In our case, of ANCA-negative EGPA, histopathological study of a bullous lesion demonstrated severe dermal oedema leading to formation of a subepidermal blister, abundant dermal eosinophils, with eosinophilic granules forming a flame figure around collagen fibrils and several non-necrotizing granulomas.
It has been hypothesized that the myriad clinical manifestations of ANCA-negative EGPA (i.e. perimyocarditis, peripheral neuropathy) might be due mainly to the eosinophilic infiltrates, whereas in ANCA-positive EGPA, the necrotizing vasculitis component might prevail (7).
In concordance with his ANCA-negative status, our patient had a dramatic clinical appearance (perimyocarditis) in association with the bullous skin eruption and histopathology of an abundant dermal eosinophil infiltrate and flame figures as an evidence of eosinophil activation and degranulation. The inflammatory infiltrate included only few neutrophils with no evidence of vasculitis. Furthermore, the fluctuation in eosinophil counts paralleled the clinical course; both cutaneous and extracutaneous.
In the absence of positive ANCA serology, known to mediate vascular endothelial injury (8), we hypothesize that bulla formation may be due to the release of eosinophilic granules (containing major basic proteins, eosinophil cationic proteins, and eosinophil-derived neurotoxin) (8), and the severe dermal oedema caused by eosinophilic dermal infiltrate. The major contributory role of the immune-complex mediated inflammation and neutrophil vascular infiltration seems less central in the pathogenesis of ANCA-negative EGPA.
Even though it is well established that the mainstay of management in EGPA is corticosteroids, with a good response and a 90–97% 5-year survival rate (8), prolonged treatment is usually required. There is currently no specification of the response of the bullous eruption. Our patient exhibited a rapid response and resolution of the eruption, with no evidence of recurrence in more than 6 months of follow-up.
In conclusion, EGPA, especially the ANCA-negative form, may manifest as a bullous phenotype exhibiting specific histopathological findings. This variant may respond rapidly to systemic corticosteroids.


 Click to show fullsize
Click to show fullsize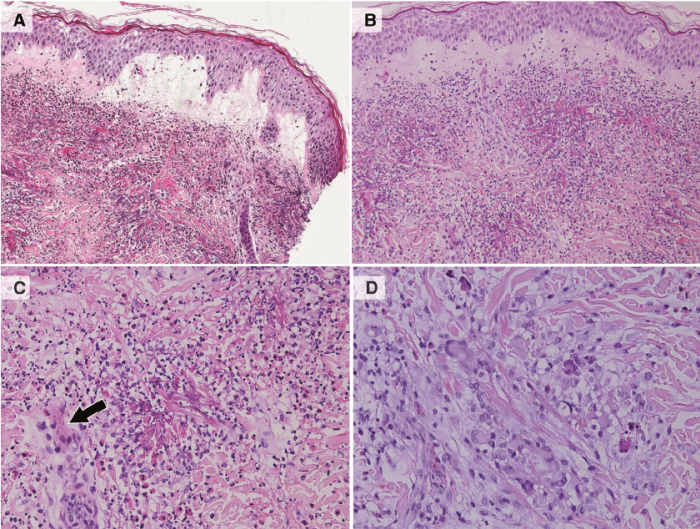 Click to show fullsize
Click to show fullsize