


1Plastic and Reconstructive Surgery Research, Stopford Building, Manchester, 2Centre for Tissue Injury and Repair, Institute of Inflammation and Repair, 3Centre for Dermatology Research, Institute of Inflammation and Repair, University of Manchester, Manchester, UK, and 4Department of Dermatology, University of Münster, Münster, Germany
Keloid disease is a fibroproliferative tumour characterised by aggressive local invasion, evident from a clinically and histologically active migrating margin. During combined laser capture microdissection and microarray analysis-based in situ gene expression profiling, we identified upregulation of the polypeptide growth factor neuregulin-1 (NRG1) and ErbB2 oncogene in keloid margin dermis, leading to the hypothesis that NRG1 contributed to keloid margin migration through ErbB2-mediated signalling. The aim of this study was to probe this hypothesis through functional in vitro studies. Exogenous NRG1 addition to keloid and normal skin fibroblasts altered cytokine expression profiles, significantly increased in vitro migration and keloid fibroblast Src and protein tyrosine kinase 2 (PTK2/FAK) gene expression. ErbB2 siRNA knockdown attenuated both keloid fibroblast migration and Src/PTK2 expression, which were not recovered following NRG1 administration, suggesting the NRG1/ErbB2/Src/PTK2 signaling pathway may be a novel regulator of keloid fibroblast migration, and representing a potential new therapeutic target.
Key words: keloid disease; laser capture microdissection; neuregulin-1; ErbB2; migration.
Accepted Nov 23, 2016; Epub ahead of print Nov 24, 2016
Acta Derm Venereol 2017; 97: XX–XX.
Corr: Dr Ardeshir Bayat, Plastic & Reconstructive Surgery Research, Institute of Inflammation & Repair, University of Manchester, Stopford Building, Manchester M13 9PT, UK. E-mail: Ardeshir.Bayat@manchester.ac.uk
Representing the extreme end of the cutaneous scarring spectrum, keloid disease is a fibroproliferative tumour characterised by excess extracellular matrix (ECM) deposition, increased inflammatory cytokine expression and aggressive local invasion (1). Although keloid is often referred to as benign, in that it does not metastasize, it behaves in a tumorigenic fashion by actively invading adjacent normal skin and spreading beyond the boundaries of the original wound (2, 3). To date, although significant progress has been made, the mechanisms underlying the migration required for keloid invasion have not been fully elucidated. The invasion and spread of keloid scars represent a significant clinical challenge not currently controlled with available therapies (4, 5).
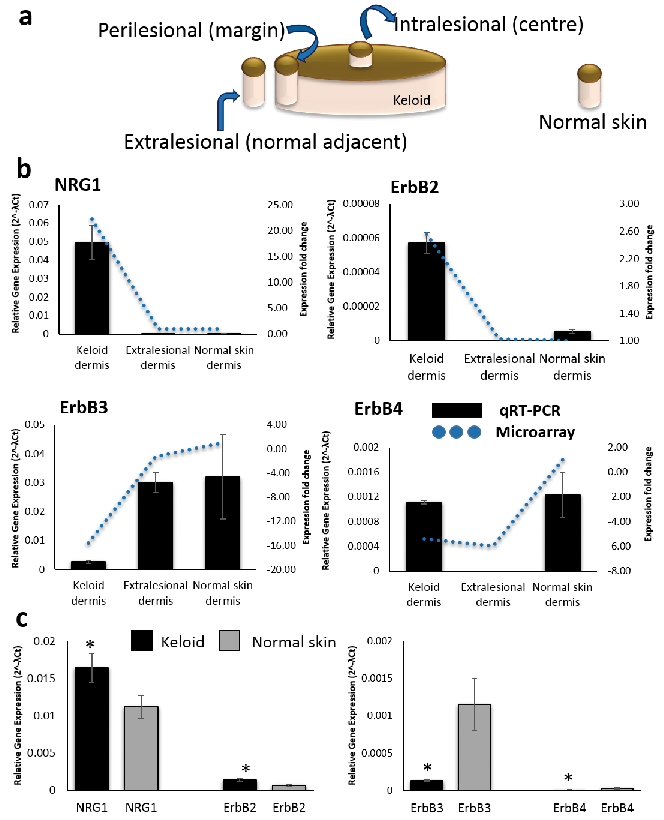
To embrace this challenge and address the concept of keloid migration, we pursued in-house microarray evidence from specific keloid tissue compartments demonstrating involvement of neuregulin-1 (NRG1) and ErbB2 (HER-2/Neu). The microarray data encompassed differential gene expression between intralesional (centre), perilesional (margin) and extralesional in situ keloid and normal skin dermis (Fig. 1a), which was achieved using laser capture microdissection (LCM). Support for our focus on the margin as the active-site of keloid migration arises from a clinically raised erythematous border, histological evidence of a hyper-cellular advancing tongue in the papillary keloid margin dermis (KMD) (6) and increased migration and collagen in fibroblasts from the growing margin of the keloid scar (7, 8). NRG1 is a polypeptide growth factor upregulated in several human cancers (9–11), is secreted from fibroblasts (12) and plays a role in skin pigmentation (13), keratinocyte migration in wound healing (14), hypertrophic scarring (15) and fibrosis (16). NRG1 binds either ErbB3 or ErbB4 resulting in recruitment and heterodimerisation with ErbB2, an orphan receptor with no affinity ligand of its own (17) but which is implicated in tumorigenesis (18, 19).
Fig. 1. Neuregulin–1 (NRG1) and ErbB2 gene expression for in situ keloid tissue and keloid fibroblasts compared with normal skin. (A) Schematic diagram of keloid biopsy sites for combined laser capture microdissection and microarray approach. Biopsies were harvested from the centre (intralesional), margin (perilesional) and keloid-adjacent normal skin (extralesional). (B) qRT-PCR (bar graph) validation of microarray data (line graph represents microarray fold change) for NRG1, ErbB2, ErbB3 and ErbB4 for in situ keloid vs normal skin. (C) qRT-PCR bar graph of keloid fibroblast and normal skin fibroblasts (n = 5) for NRG1, ErbB2, ErbB3 and ErbB4. Data are mean ± SEM from 3 independent experiments (*p < 0.05).
The functional association between NRG1 and ErbB2 on cell migration has been observed in cardiac myocytes, Schwann cells and glioma tissue, indicating the NRG-1β/ErbB-dependent activation of Src/FAK modulates cell–cell contact, cell motility and focal adhesion complex formation (20, 22). Indeed protein tyrosine kinase 2 (PTK2/FAK), a non-receptor protein tyrosine kinase, was shown to be upregulated in keloid disease, where it influences cell migration through alteration of the focal-complex assembly disassembly cycle (23–25). The aggressive invasion evident in keloid disease supports the hypothesis of a mechanobiological aetiology (26, 27), where rapid focal adhesion turnover and modulation of tension actin filaments produce a “migration-related disease” (28). Our site-specific approach localised upregulation of NRG1 and ErbB2 to the isolated KMD, leading to our hypothesis that NRG1 overexpression promotes fibrosis and fibroblast migration into adjacent normal skin through ErbB2-mediated signaling.
In this study, we confirmed our microarray finding of NRG1 and ErbB2 overexpression in KMD compared with normal skin on both gene and protein levels. We then sought to demonstrate the pro-fibrotic and pro-migratory effects of NRG1 and to establish whether these effects are dependent on ErbB2 upregulation. We showed that keloid fibroblast expression reflects that of the KMD, supporting the use of functional in vitro assays to test our hypothesis. Exogenous NRG1 administration resulted in upregulation of pro-fibrotic factors, Src and PTK2 as well as increased fibroblast migration on in vitro scratch assay. The use of ErbB2 siRNA attenuated this migration and the expression of Src and PTK2 in keloid fibroblast. Failure to recover this expression with re-introduction of NRG1 suggested the effects were mediated through ErbB2.
Keloid (centre, margin and extralesional) (Fig. 1a) and normal skin biopsies were harvested according to Declaration of Helsinki protocols with full written consent (North West Research Ethics Committee Ref. 11/NW/0683). Biopsies were stored in RNA stabilisation solution then OCT-embedded for serial cryosection or formalin-fixed and paraffin-embedded (FFPE). Demographic data are detailed in Table SI, which indicates that some of the keloid and normal skin samples differ in terms of ethnicity and prior treatment.
To preserve RNA integrity, a rapid staining protocol was used (LCM Staining Kit, Ambion, USA) prior to laser capture microdissection (PALM, Carl Zeiss, MicroBeam 4.2, Germany). The LCM, mRNA extraction, amplification and microarray analysis were performed as described previously (29).
Performed on mRNA from laser-captured elements and cultured fibroblasts, as previously described (29). Primer lists detailed in Table SII. Reactions were performed in triplicate and normalised against two house-keeping genes, RPL32 and GAPDH.
Primary fibroblast culture was established using standard techniques as described (8). In brief, tissue was minced, incubated in collagenase, combined with complete DMEM for 3 h at 37°C. Cells were pelleted and grown in tissue culture flasks with medium changes every 48–72hrs until confluent. Passages 1–3 were used.
Normal skin and keloid fibroblast were seeded into 6-well plates in complete DMEM. Once confluent, cells were serum-starved for 24 h before adding 50 ng/ml recombinant human neuregulin-β1 (rhNRG1-β1) (PeproTech, UK) for 24 h. Control cells were treated with serum-free medium for a total of 48 h. Cells were harvested using Trizol (Life Technologies, UK) for RNA extraction (RNeasy Mini Kit, Qiagen) or radioimmunoprecipitation assay buffer (RIPA, Sigma-Aldrich) for western blot analysis.
To determine ErbB dimerisation, co-immunprecipitation of ErbB receptor complexes was performed according to the manufacturer’s protocol (Pierce co-immunoprecipitation (Co-IP) kit, ThermoFisher Scientific, UK). This is discussed in detail in Appendix S1 and the antibody list detailed in Table SIII. Blots were developed using nitro blue tetrazolium/S-bromo-4-chloroindoxyl phosphate (NBT/BCIP, 34042, ThermoFisher Scientific, UK).
FFPE sections (5 µm) were stained for ErbB2 using manufacturer’s instructions for Novolink® peroxidase detection kit (Leica Biosystems, UK) and red chromogen Vector® NovaRED™ substrate kit (Vector, CA, USA) to differentiate from melanin then counterstained with haematoxylin. Combined ErbB2/NRG1 immunofluorescence was performed using citrate buffer (pH 6) antigen retrieval for 20 min at 90°C. A negative control was performed in each without primary antibody. (Antibody incubation details Table SIII).
Following optimisation, 5 nM ErbB2 siRNA (Silencer® select s613 validated, ThermoFisher scientific UK) or control siRNA (Silencer® Select Negative Control No. 1, ThermoFisher scientific UK) was introduced to keloid fibroblast using Lipofectamine® RNAiMAX (Life Technologies, UK) according to manufacturer’s instructions. Culture medium (Opti-MEM I, ThermoFisher Scientific, UK) for RNAi transfection was antibiotic and serum-free. Cells were harvested for RNA extraction or protein analysis by western blot.
This is described in detail in the Appendix S1. In brief, fibroblasts were seeded into 6 well plates and a scratch introduced with a 10 µl pipette tip. Cells were then treated with either rhNRG1, ErbB2 siRNA or both rhNRG1 and ErbB2 siRNA together. Images were taken at time 0 and 48 h and compared to quantify the number of migrated cells which were then presented as mean ± standard error of mean (SEM).
1x104 keloid and normal skin fibroblast seeded per well into 96-well plates and serum-starved for experiment duration. Cells were treated with 50 ng/ml rhNRG1 for 72 h, 5 nM ErbB2 siRNA or control siRNA as indicated. Controls were treated with serum-free medium alone. Metabolically active cell number was assessed using MTT assay as described previously (30). Absorbance (550–690 nm) was measured (FLUOstar Optima, BMG Labtech, UK) and expressed as mean ± SEM from 3 independent experiments.
For qRT-PCR, gene expression levels were normalised against two internal controls and ΔΔCT calculated. All data are represented as mean ± SEM. Statistical analysis was calculated using Student’s t test and one way ANOVA with Tukey post-hoc correction (SPSS, IBM), where p-value < 0.05 considered statistically significant.
Combined LCM and microarray allowed site-specific comparison of keloid centre, margin and adjacent lesional sites with normal skin for epidermis and dermis (Fig. 1a). NRG1 was identified as most significantly upregulated in KMD (fold change 22.24, p-value 1.59E-05) but was also upregulated in keloid centre dermis (fold change 10.71, p-value 0.0002). Given the upregulation of NRG1, we examined the microarray data for potential interaction candidates using criteria of fold change > 2 and p-value < 0.05. ErbB2 was identified as most significantly upregulated in KMD (fold change 2.56, p-value 0.0031). ErbB3 (fold change 15.6, p-value 6.82E-07) and ErbB4 (fold change 5.42, p-value 0.0145), however, were significantly downregulated in KMD. Findings were validated using qRT-PCR, which reflected the microarray expression pattern (Fig. 1b). Full expression data is detailed in Table SIV. Additionally, qRT-PCR (n = 5) confirmed keloid fibroblast expressed increased NRG1α (p = 0.044) and NRG1β (p = 2.7E-05) as well as ErbB2 (p = 0.011) with reduced expression of ErbB3 (p = 0.009) and ErbB4 (p = 0.034) compared with normal skin fibroblast (Fig. 1c). Therefore, keloid fibroblast in culture reflect the NRG1/ErbB gene expression of in situ keloid dermis, which is the presumed site leading keloid migration (31, 32). Interestingly, the strength of our LCM approach was demonstrated in the accuracy of in situ differential gene expression between keloid and normal skin compared with monolayer culture, which minimised the degree of gene upregulation in keloid versus normal skin dermis. However, given the complex nature of keloid disease and the fact that there is no validated animal model for keloid disease (33), the in vitro experiments are essential to confirm in situ observations made in human keloid disease tissue in order to progress towards mechanistic understanding of the pathogenesis.
To assess whether gene expression findings translated into protein changes, we performed immunohistochemistry for ErbB2 (Fig. 2a, b) and immunofluorescence for combined NRG1 and ErbB2 (Fig. 2a). 89% of keloid dermis samples showed increased ErbB2 expression (n = 9) compared with normal skin (n = 5), where there was no dermal expression. Interestingly, positive ErbB2 expression in keloid dermis was associated with histological features considered to be pathognomonic of keloid disease (6), including dermal nodule (Fig. 2bi), horizontal fibrous band (Fig. 2bii), obliteration of papillary/reticular border (Fig. 2biii) and papillary dermal inflammatory infiltrate (Fig. 2biv). Combined NRG1/ErbB2 immunofluorescent staining (yellow stain) demonstrated co-localisation within the dermis (Fig. 2a), which was increased in keloid (n = 13) compared with normal skin (n = 6). Similar to ErbB2 immunohistochemistry, this co-localised expression in keloid dermis was more frequently in areas of inflammatory infiltrate. When only KMD sections were considered, both NRG1 and ErbB2 were positive in 6/6 examined patients, supporting microarray data.
Fig. 2. Neuregulin–1 (NRG1) and ErbB2 protein expression in keloid versus normal skin (NS). (A) ErbB2 immunohistochemistry for KD (n = 9) and NS (n = 5) show ErbB2 positive stain (black arrows) in KD sections only. Combined NRG1 (green/FITC) ERbB2 (red/TRITC) immunofluorescence for KD (n = 13) and NS (n = 6). Dermal co-localisation (white arrows indicating yellow stain) increased in KD vs NS. Negative control with no primary antibody treatment shown also. (B) ErbB2 immunohistochemistry of keloid margin (n = 6). Positive staining associated with i) hypercellular dermal nodule ii) horizontal fibrous band iii) papillary-reticular border obliteration iv) papillary dermis (PD) inflammatory infiltrate. (C) Western blot and mean fold change quantification for NRG1 and ErbB2 in keloid fibroblasts and NS fibroblasts (where mean fold change is the average of n = 5 after each is divided by its own control (GAPDH). Scale bar = 50 µm. Scale bar of inset magnification = 10 µm. RD, reticular dermis.
Protein expression, determined by western blot (n = 5), showed a trend toward increased expression of both NRG1 and ErbB2 (Fig. 2c) with decreased ErbB3 and ErbB4 expression in keloid fibroblast compared with normal skin fibroblast (Fig. S1). Overall, our results show increased NRG1 and ErbB2 protein expression in keloid dermis compared with normal skin, supporting gene expression findings.
To evaluate ErbB dimerisation status, co-immunoprecipitation was performed using keloid and normal skin fibroblast lysates (n = 3) and the immunoprecipitate subjected to western blot analysis (Fig. 3). Keloid fibroblast immunoprecipitation of ErbB2 led to increased co-precipitation of ErBB2 (p = 0.003) and ErbB3 when compared with normal skin fibroblast. Additionally, ErbB3 and ErbB4 homodimers preferentially occurred in normal skin versus keloid fibroblast. Assay specificity was demonstrated by absence of immunoreactivity in negative controls. These results suggest keloid fibroblasts express higher levels of ErbB2/ErbB2 homodimers and ErbB2/ErbB3 heterodimers than normal skin fibroblasts.
Fig. 3. Co-immunoprecipitation of total keloid and normal skin fibroblast protein lysate (n = 3) and western blot analysis with antibody against ERbB2, ErbB3 and ErbB4. The immunoprecipitate (IP) pull down and associated western blot (WB) for each specific antibody probed shown with the accompanying quantification graph under the relevant headings. Quantification was performed with ImageJ analysis and this was repeated three times. Data are represented as mean ± SEM where *p < 0.05 using Student’s t test. The western blots show each sample for n = 3 for both keloid fibroblast and normal skin fibroblast. The patient lanes are marked with “P” and the negative control lanes for that same patient marked with “N”.
As the more active isoform, exogenous rhNRG1-β1 was added to normal skin fibroblasts at 50 ng/ml for 24 h and compared with untreated (serum-free medium) normal skin fibroblasts (n = 5). qRT-PCR was performed for a number of pro-fibrotic genes known to be dysregulated in keloid disease (1, 34–37) (Fig. 4a, b). Treatment of normal fibroblasts with rhNRG1 resulted in significant upregulation of collagen I (p = 0.0016), collagen III (p = 0.001) and fibronectin (p = 0.007) as well as pro-inflammatory cytokines transforming growth factor receptor (TGFβ1) (p = 0.04), IL-6 (p = 0.0059) and IL-8 (p = 0.0019). These results indicate exposure to NRG1 produces a pro-fibrotic response in normal skin fibroblasts that reflects the keloid expression profile.
Fig. 4. Fibroblast (n = 5) treatment with recombinant human neuregulin–1 (rhNRG1). (a) qRT-PCR showing significantly increased collagen I, collagen III, fibronectin (FN), IL-6 and IL-8 expression in rhNRG1-treated normal skin fibroblasts (NSF) and (b) significantly increased transforming growth factor (TGFβ1) in keloid fibroblasts (KF) and NSF. (c) Representative micrographs of in vitro scratch migration assay (n = 3) showing KF/NSF response towards injury at 0 h and 48 h with/without rhNRG1 treatment. (d) Average number of migrated KF/NSF at 48 h were counted. (e) qRT-PCR for Src, FAK/PTK2 and signal transducer and activator of transcription (STAT3) of rhNRG1-treated vs untreated KF/NSF. Data are mean ± SEM from at least 3 independent experiments (*p < 0.05, **p < 0.005).
Fig. 4b shows keloid fibroblast treatment with 50 ng/ml rhNRG1 for 24 h (n = 5) stimulated significant upregulation of TGFβ1 (p = 3.97E-07). There was also upregulation of fibroblast growth factor (FGF7) (34) (p = 0.0086) (Fig. S2) as well as a trend toward increased expression of collagen I, collagen III, fibronectin and IL-6 (Figs S3 and S4).
In vitro scratch assay was used to assess migration capacity of keloid and normal skin fibroblasts (Fig. 4c). Untreated fibroblasts were compared to those stimulated with 50 ng/ml rhNRG1 over 48 h. Untreated keloid fibroblast showed significantly more migration than normal skin fibroblast. Additionally, rhNRG1 treatment resulted in significantly increased migration for keloid fibroblasts (p = 0.02) and normal skin fibroblasts (p = 5.0E-05) over untreated cells (Fig. 4d). A 72-h MTT assay (n = 4) on all fibroblasts confirmed cell numbers remained constant for experimental duration and were equal between keloid and normal skin fibroblasts (Fig. S5).
Given that NRG1 is known to affect cell migration and motility through effects on Src/FAK and signal transducer and activator of transcription (STAT3) (21, 37–40), here we investigated the effect of rhNRG1 on these molecules in keloid and normal skin fibroblasts. Fig. 4f demonstrates qRT-PCR of keloid fibroblasts (n = 5) treated with rhNRG1 showed significant upregulation of Src (p = 0.006), PTK2/FAK (p = 0.04) and STAT3 (p = 0.005).Normal skin fibroblasts (n = 5) also showed significant upregulation of Src (p = 0.016) and STAT3 (p = 0.004) but not PTK2.
To clarify ErbB2 involvement in keloid fibrosis and migration, keloid fibroblasts (n = 5) were treated with 5nM ErbB2 siRNA or scrambled siRNA negative control for 48 h and successful knockdown confirmed by western blot (Fig. 5a). qRT-PCR of ErbB2 knockdown keloid fibroblasts showed significantly less expression of collagen I (p = 1E-05), collagen III (p = 0.019), fibronectin (p = 0.012) and TGFβ1 (p = 0.04) than negative control (Figs S6 and S7).
Fig. 5. ErbB2 siRNA-transfected keloid fibroblasts (KF). (a) Quantification (n = 5) and western blot showing significant (~90%) ErbB2 protein (30 µg) expression reduction in siRNA-transfected (red arrow) vs untreated (blue arrow) and negative control (black arrow) KF. (b) 72 h MTT of keloid fibroblasts (KF). (c) Average number of migrated KF plotted on the graph following the indicated treatments. (d) Representative micrographs of in vitro scratch migration assay (n = 3). Fibroblast migration response towards injury at 48 h following treatment: negative control, ErbB2 siRNA or both ErbB2 siRNA and neuregulin (NRG1) together. (e) qRT-PCR for PTK2, Src and STAT3 expression of KF treated with negative control, ErbB2 siRNA or both ErbB2 siRNA and rhNRG1 together. Data are mean ± SEM from at least three independent experiments (*p < 0.05, **p < 0.005).
The MTT assay (n = 4) of siRNA-treated keloid fibroblasts confirmed viability was maintained over the 72 h experimental period with minimal cell proliferation in serum-free environment, as expected (Fig. 5b). Therefore, the significantly reduced cell migration seen in ErbB2 knockdown keloid fibroblasts (p = 0.005) was not due to apoptosis (Fig. 5c, d). Reduced migration of ErbB2 siRNA-transfected keloid fibroblasts, seen with in vitro scratch assay, is supported by significantly reduced gene expression of PTK2 (p = 0.0002) and Src (p = 0.003) compared with negative control (Fig. 5e).
Having demonstrated the effect of ErbB2 knockdown on keloid fibroblast migration, we next sought to determine whether the observed pro-migratory effects of NRG1 on keloid fibroblasts were mediated through ErbB2. Addition of rhNRG1 to keloid fibroblasts transfected with ErbB2 siRNA did not result in a significant increase in migration (p = 0.29) on in vitro scratch assay (Fig. 5c, d). Nor did addition of rhNRG1 to ErbB2 knockdown keloid fibroblasts restore the expression of PTK2 or Src (Fig. 5e), suggesting effects of NRG1 in keloid disease are mediated by ErbB2 overexpression and other ErbB members are unable to compensate for ErbB2 loss or they may use alternate signaling mechanisms.
Keloid disease is a quasineoplastic cutaneous tumour characterised by migration into surrounding skin beyond the original wound borders, failure to regress and high recurrence rates following excision (1). Our identification of NRG1 and ErbB2 upregulation in the clinically and histologically active (6, 41) KMD in situ, suggested a heretofore unexamined mechanism underlying invasive migration contributing to keloid disease pathogenesis.
Here, we have demonstrated keloid fibroblast reflect in situ KMD tissue, with increased NRG1 and ErbB2 but decreased ErbB3 and ErbB4 expression compared to normal skin. ErbB2 overexpression leads to homodimerisation and constitutive activity through autophosphorylation, leading to ligand-independent signaling (42, 43). Alternatively, ErbB2 is the preferred dimerization partner of both ErbB4 and kinase-impaired ErbB3 (44, 45), which even at low levels can produce potent signal transduction through ErbB2/ErbB3 heterodimerisation (46). Given heterodimerisation with ErbB2 generates the most robust signaling downstream, we hypothesised the downregulation of ErbB3 and ErbB4 discouraged their homodimerisation in favour of ErbB2 (47), achieving maximal signaling activity with NRG1 affinity (48) (Fig. S8). This is supported by our finding of increased ErbB2 homodimers and ErbB2/ErbB3 heterodimers in the co-immunoprecipitation of keloid versus normal skin fibroblasts. Although ErbB3 and ErbB4 serve as the NRG1 receptors, we have shown that ErbB2 ablation hinders keloid cell’s ability to respond to NRG1 (18).
This study has shown that rhNRG1 treatment elicited a keloid-like pro-fibrotic cytokine expression profile in normal skin fibroblasts, with significant upregulation of collagen I, collagen III, TGFβ1, fibronectin, IL-6 and IL-8. These observations led us to hypothesise a possible mechanism whereby paracrine NRG1 signals from migrating/advancing keloid fibroblast may alter the expression profile of adjacent normal skin fibroblast, thereby driving keloid expansion at the margin into extralesional/normal skin (Fig. S9) (12). NRG1 was recently found to be upregulated in hypertrophic scar fibroblasts, where it regulated ECM expression through connective tissue growth factor (CTGF) (15). Here, although we found exogenous rhNRG1 treatment resulted in decreased CTGF expression in keloid fibroblast (Figs S10 and S11), this does not detract from previous findings in hypertrophic scarring. Indeed, these differences may have clinical implications given the continued difficulties with differential diagnosis between keloid and hypertrophic scars. However, further studies are required to explore the mechanisms underlying these divergent responses to rhNRG1 in vitro.
We demonstrated reinforcement of TGFβ1 overexpression in rhNRG1-treated keloid fibroblasts. TGFβ1, a pleiotropic growth factor already known to be upregulated in keloid disease (1, 36), has been postulated to facilitate scar expansion through pro-inflammatory and pro-migratory effects (49, 50). Interestingly, our finding of NRG1 and ErbB2 co-localisation in keloid dermis was frequently observed within previously described areas of keloid inflammatory infiltrate (51). TGFβ1 is known to influence cell migration through MMP regulation (52) and its effect on single cell motility is thought to require synergy with the EGFR family of tyrosine kinase receptors (53, 54), such as ErbB2 (55–57). Both ErbB2 and TGFβ also independently activate PI3K/Akt (58, 59) and MAPK/Erk (60, 61) pathways, which are regulators of migration and are both known to be dysregulated in keloid disease (62, 63).
Our finding of TGFβ1 upregulation and CTGF downregulation following rhNRG1 treatment of keloid fibroblasts is curious, especially as TGFβ is well known to induce CTGF in keloid fibroblasts (59). Thus, NRG1 upregulation may only be one contributing element in the regulatory systems that underlie increased TGFβ1 in keloid disease (3); in combination with other factors inducing TGFβ1 overexpression, this may override the negative effects of NRG1 on the CTGF expression seen here. Alternatively, the incompletely understood complexity of Smad/non-Smad signaling, as well as the other signaling regulators that are known to affect CTGF expression and to be dysregulated in keloid disease including mechanical stress/tension (1, 64–66), may drive the overall expression of CTGF in keloid disease in vivo. While this is an interesting finding and represents a focus for future research, further investigation into this relationship was beyond the scope of the current study.
NRG1 adheres to ECM in vivo, thereby activating ErbB2 receptors on invading cells and contributing to cell migration (20). Here, we show rhNRG1 stimulation promoted significant cell migration in both normal skin and keloid fibroblast. Additionally, we demonstrated rhNRG1 treatment resulted in significant upregulation of Src in normal skin fibroblast and both Src and PTK2 in keloid fibroblast. Importantly, the small but significant upregulation of PTK2 in concert with Src, seen only in keloid fibroblast and which is considered essential to the establishment of an invasive cell phenotype (67, 68), may contribute to the migratory fibroblast phenotype that differentiates keloids from normal scars. We established the role of ErbB2 in this study through ErbB2 siRNA studies, which resulted in significant reduction in both cell migration in vitro and also downregulation of Src and PTK2 gene expression in keloid fibroblast. PTK2 facilitates migration through ECM-integrin junction signaling (69), also playing a role in inflammation and fibrosis (70). The site-specific approach in our study highlights identification of ErbB2 and NRG1 dysregulation in keloid dermis as opposed to epidermis and interestingly, dermal PTK2 has been suggested as more vital to cutaneous healing than epidermal (70).
To determine whether the effects of NRG1 and ErbB2 on migration were co-dependent, we treated the ErbB2 knockdown keloid fibroblast with rhNRG1. This treatment failed to significantly increase in vitro migration or gene expression of Src and PTK2 in keloid fibroblasts, indicating NRG1 requires ErbB2 for its pro-migratory effects. STAT3 has been previously implicated in keloid migration (71) and it has been suggested that increased ErbB2 can activate STAT3 via Src (72, 73). Interestingly, our results revealed a significant STAT3 increase (p < 0.01) in both normal skin and keloid fibroblasts treated with rhNRG1. However, ErbB2 knockdown in keloid fibroblasts did not significantly reduce STAT3 or known STAT3 modulator IL-6 when compared with untreated keloid fibroblast controls (74–76). Additionally, STAT3 in KMD in our microarray data was marginally downregulated (fold change 2.02, p-value 0.0051), suggesting NRG1 is not the dominant regulator of STAT3 and any stimulation of these molecules may contribute to fibroblast migration through alternative mechanisms (77). Although STAT3 has been shown to play a significant role in keloid pathogenesis, the site-specific nature of this study may account for differences in observed expression (78).
One of the limitations of our study was the availability of optimal keloid tissue samples with few of the keloid samples having previous treatment and only some of the keloid patients were Caucasian, whereas all of the normal skin samples were from Caucasian patients. Given the ethical implications of excising normal skin tissue from keloid-prone patients distant to the scar, we matched the samples to the maximum degree within our control. Although not ideal, the extralesional tissue may be interpreted as a type-matched control and in the extralesional dermis, NRG1, ErbB2 and ErbB3 were not dysregulated (Table SIV).
In summary, we have shown NRG1 and ErbB2 upregulation in KMD and that suppression of ErbB2 expression in keloid fibroblasts neutralises the pro-fibrotic cytokine expression profile and pro-migratory effects of NRG1. This study examines the molecular mechanisms underlying keloid migration, which is an essential component of the invasion that is pathognomonic of keloid disease. The upregulation of NRG1 and ErbB2 may promote migration through effects on JAK/STAT signalling, TGFβ and/or Src/FAK. Confirmation of the functional importance of these findings through keloid organ culture may provide further evidence supporting the exposure in this study of a potential role for NRG1/ErbB2/FAK/Src signaling localised to KMD, which presents an intriguing mechanistic aspect of keloid migration not previously investigated. The potential future clinical implications include current availability of targeted therapies for ErbB2, which with site-specific application may be a novel strategy in the management of keloid disease.
The authors are grateful to Yaron Har-Shai and Guyan Arscott for their assistance with sample provision and to Adam Taylor for his technical assistance with analysis.
The authors declare no conflict of interest.
 Appendix S1 Figure SI Figure S2 Figure S3 Figure S4 Figure S5 Figure S6 Figure S7 Figure S8 Figure S9 Figure S10 Figure S11 Table SI Table SII Table SIII Table SIV
Appendix S1 Figure SI Figure S2 Figure S3 Figure S4 Figure S5 Figure S6 Figure S7 Figure S8 Figure S9 Figure S10 Figure S11 Table SI Table SII Table SIII Table SIV

 Click to show fullsize
Click to show fullsize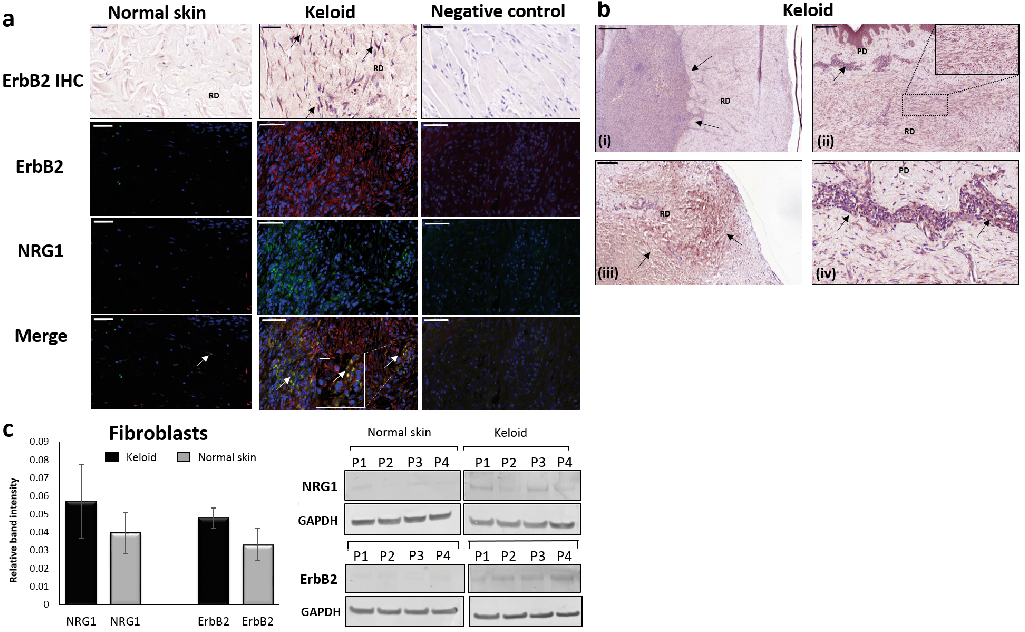 Click to show fullsize
Click to show fullsize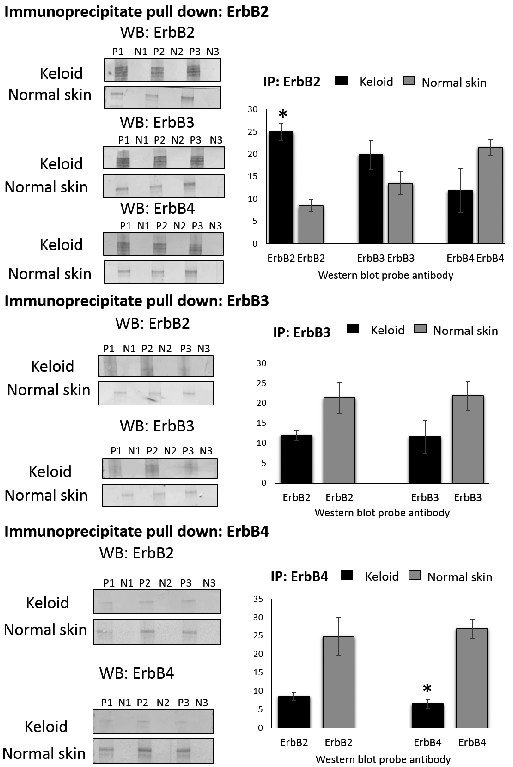 Click to show fullsize
Click to show fullsize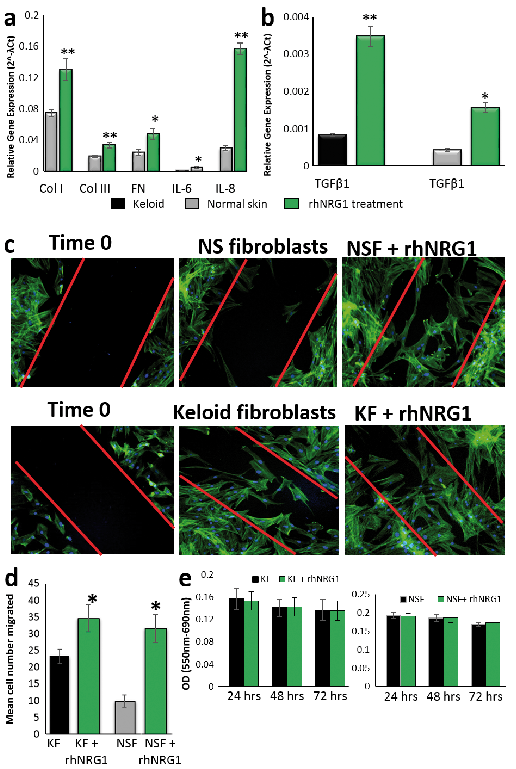 Click to show fullsize
Click to show fullsize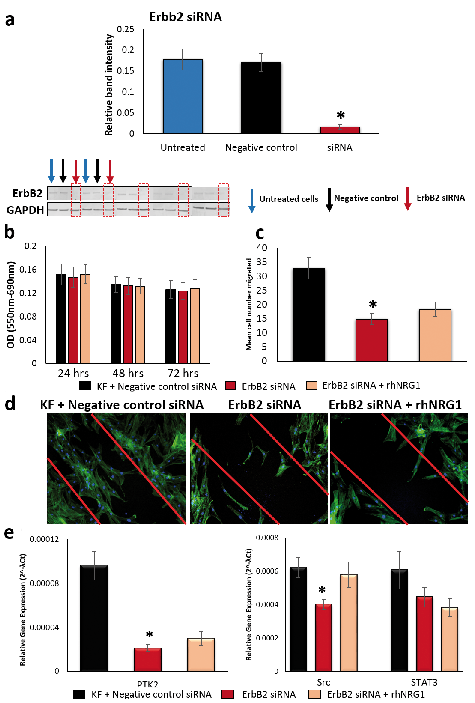 Click to show fullsize
Click to show fullsize