


Departments of 1Dermatology and 2Hematology, Hokkaido University Graduate School of Medicine, North 15 West 7, Kita-ku, Sapporo 060-8638, Japan. *E-mail: h-ujiie@med.hokudai.ac.jp
Accepted Nov 23, 2016; Epub ahead of print Nov 24, 2016
Linear IgA bullous dermatosis (LABD) is a rare subepidermal autoimmune blistering disorder in which most cases have IgA autoantibodies to the 120-kDa and/or 97-kDa shed ectodomains of type XVII collagen (COL17, BP180) at the basement membrane zone (BMZ) (1). The aetiology of LABD remains largely unclear, although associations with drugs, infections and malignancies have been reported (2). Immunoglobulin light-chain (AL) amyloidosis is caused by the deposition, in tissue, of misfolded light-chains, which are produced by clonal plasma cells, resulting in multi-organ dysfunction (3). AL amyloidosis often develops against a background of monoclonal gammopathy. It has been reported that some monoclonal antibodies from patients with monoclonal gammopathy possess antigen-binding activity directed to autoantigens (4).
We report here a case of LABD associated with IgA AL amyloidosis. To the best of our knowledge, this is the first case of LABD associated with systemic AL amyloidosis.
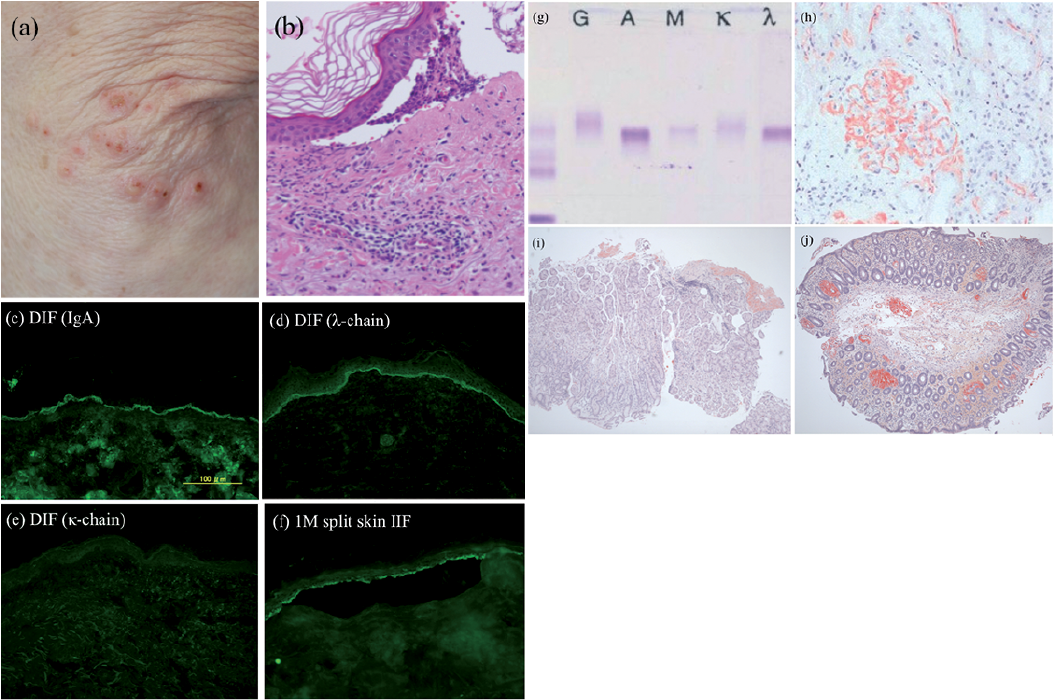
A 73-year-old Japanese man was referred to our outpatient clinic with a 1-week history of vesicles on the extremities. He had also had severe proteinuria and hypertension for 6 months and had received a thorough examination at the department of haematology. He had taken no additional medications for one year before the onset of bullous lesions. Physical examination revealed numerous tense vesicles less than 5 mm in diameter surrounded by itchy erythema on the left elbow and the ankles and lower back (Fig. 1a). A biopsy specimen obtained from a vesicle on the left elbow showed a subepidermal blister, which contained neutrophils and eosinophils (Fig. 1b). Direct immunofluorescence (DIF) demonstrated linear deposits of IgA (Fig. 1c), but not of IgG or IgM, at the BMZ. Further investigation revealed that those IgA autoantibodies possess the lambda-light chain (Fig. 1d), but not the kappa-light chain (Fig. 1e). Indirect immunofluorescence (IIF) was positive for IgA at the BMZ up to ×64 dilutions. 1M NaCl-split skin IIF was positive for IgA on the epidermal side (Fig. 1f). Chemiluminescent enzyme immunoassay of COL17 NC16A was negative for IgG. From these findings, a diagnosis of LABD associated with IgA-lambda autoantibodies was made.
Fig. 1. Clinical and analytical characteristics of the patient. (a) Tense vesicles surrounded by itchy erythema on the left elbow. (b) Biopsy specimen from a vesicle on the left elbow shows subepidermal blisters containing numerous eosinophils and neutrophils (haematoxylin and eosin staining, original magnification ×100). (c–e) Direct immunofluorescence (DIF) reveals linear deposits of (c) IgA and (d) lambda light chain, but not (e) kappa light chain at the BMZ of lesional skin. (f) 1M NaCl-split skin IIF is positive for IgA on the epidermal side. (g) Immunofixation reveals monoclonal paraprotein IgA-lambda in the serum. (h) Mesangial deposits of amyloid on the renal specimen (direct fast scarlet (DFS) staining, original magnification ×200). Deposits of amyloid on: (i) submucosa of the rectum and (j) the lamina propria and submucosa of the sigmoid colon (DFS staining, original magnification ×40).
Laboratory tests revealed kidney function to be within the normal range and showed a significantly low free light chain kappa/lambda ratio of 0.09 (normal: 0.26–1.65). Serum protein electrophoresis identified a monoclonal protein in the IgA region. Immunofixation revealed the presence of monoclonal paraprotein IgA-lambda in the serum (Fig. 1g). Urine immunofixation was negative for Bence Jones protein. Bone marrow biopsy showed 8% lambda light chain-restricted plasma cells. Direct fast scarlet (DFS) staining showed homogenous deposits of amyloid on the mesangium of a renal specimen (Fig. 1h). Mucosae in the sigmoid colon and rectum were also affected (Fig. 1i, j). Based on these results, a diagnosis of IgA-lambda type systemic AL amyloidosis with underlying monoclonal gammopathy of renal significance (MGRS) was also made. After the diagnosis, we re-checked the patient, but found no cutaneous signs of amyloidosis, such as periorbital ecchymoses or macroglossia. Direct fast scarlet (DFS) staining revealed the absence of amyloid in the patient’s skin. A combination chemotherapy with bortezomib and dexamethasone (BD therapy) was started for the treatment of AL amyloidosis. The bullous lesions disappeared and serum autoantibodies to the BMZ became undetectable by IIF within 2 weeks of initiation of treatment. Three months after initiation of BD therapy, paraprotein became negative in the serum by immunofixation and the free light chain kappa/lambda ratio became normal (0.83, normal: 0.26–1.65). Sixteen courses of BD therapy have been performed to date, and no clinical recurrence has been observed during 16-month follow-up.
To our knowledge, this is the first case of LABD concurrent with systemic AL amyloidosis. Systemic amyloidosis can cause several cutaneous and mucosal manifestations, such as purpuras, papules, plaques, oedema, macroglossia and bullous lesions that, together, are called bullous amyloidosis (BA). BA tends to appear as haemorrhagic blisters with purpura on areas of friction, suggesting that frictional trauma on regions with dermal amyloid deposits induces blister formation (5). Autoantibodies have never been detected in skin lesions or sera in cases of BA. Thus, we were easily able to distinguish LABD from BA by the immunological findings in the present case.
The mechanism of the production of autoantibodies in our case remains to be addressed. Of note, IgA-lambda was detected in serum and urine by immunofixation and at the BMZ of the skin by DIF, suggesting a correlation between LABD and systemic AL amyloidosis with MGRS in our case. MGRS is a monoclonal gammopathy that fulfils the diagnostic criteria of monoclonal gammopathy of undetermined significance (MGUS): the serum monoclonal (M) protein and bone marrow plasma cells are < 3 g/dl and 10%, respectively, and kidney disease is shown (6). In some cases of Waldenström’s macroglobulinaemia and myeloma, M proteins have been reported to have antibody activity and to react with autoantigens, such as serum albumin, thyroglobulin, nucleoprotein-associated antigens, neural antigens and cytoskeletal antigens (4). We consider the possibility that the same aberrant plasma cell clone would produce excess lambda light chains as well as intact IgA antibodies, which have the ability to react with autoantigens at the BMZ, although the possibility of incidental coexistence of LABD and AL amyloidosis cannot be excluded. As we used a secondary antibody to the Fc portion of IgA for IF studies, the IgA autoantibodies detected at the BMZ must be in an intact form. Considering that a light chain needs to be combined with a heavy chain to form a complete antigen binding site, lambda light chains detected at the BMZ would be a part of intact IgA. The immediate effect of combination chemotherapy for AL amyloidosis on LABD also indicates the relevance of these 2 disorders.
In conclusion, the present case indicates that monoclonal antibodies produced in patients with monoclonal gammopathy may possess reactivity to skin components and lead to the development of autoimmune blistering diseases.


 Click to show fullsize
Click to show fullsize