


1Department of Dermatology, Toulouse University Hospital, 24 chemin de Pouvourville TSA, FR-31059 Toulouse cedex 9, France, 2Department of Clinical Genetics, Academic Medical Center, University of Amsterdam, The Netherlands, 3Anatomo Pathology Department, IUC Oncopole, Paul Sabatier University, Toulouse, 4U 1056 INSERM – FRE 3742 CNRS - Université Toulouse III ‘Différenciation Epithéliale et Autoimmunité Rhumatoïde’, Toulouse, and 5Medical Genetics Department, CHU Purpan, Paul Sabatier University, Toulouse, France. E-mail: maella.severino@hotmail.fr
Accepted Mar 15, 2017; Epub ahead of print Mar 15, 2017
Focal dermal hypoplasia (FDH) or Goltz syndrome is an ectodermal and mesodermal disorder characterized by abnormalities of various organs including the skin. Cutaneous anomalies include skin hypoplasia, hyper- or hypo-pigmented areas following the lines of Blaschko, nodular fat herniation and papillomas. Hair can be sparse and brittle. Nail findings include ridging, dysplasia or hypoplasia. Many other organs may be involved, the most common anomalies being skeletal anomalies (especially on the extremities), short stature, coloboma of the iris and retina (1–3).
FDH is caused by mutations of the PORCN gene (Xp11.23) (4, 5) encoding a 461 amino acid 52-kDa protein involved in the secretion and signalling of WNT proteins, which plays a key role in embryonic tissue development (6). Approximately 250 individuals with FDH have been reported in the literature so far, either in small series (3–18 patients) (2) or in case reports (7). In 2016, 119 different mutations were registered in the PORCN mutation database (http://www.lovd.nl/PORCN). The majority of mutations are scattered throughout the entire coding sequence of the PORCN gene. Large deletions and splice site mutations were also reported (2). FDH is inherited as an X-linked dominant disorder, with females accounting for 90% of affected individuals. Ninety-five percent of affected women have de novo mutations, whereas 5% inherit the mutation from their parents (the mother or, rarely, the father, is affected by post-zygotic mutation) (2, 8). In men, hemizygosity for a mutation in PORCN (9) is presumed to be lethal. However, males may survive in case of Klinefelter syndrome (8) or mosaic forms (2). Postzygotic mutations of PORCN have occasionally been reported in females.
We report here 2 novel cases of FDH mosaic in female patients.
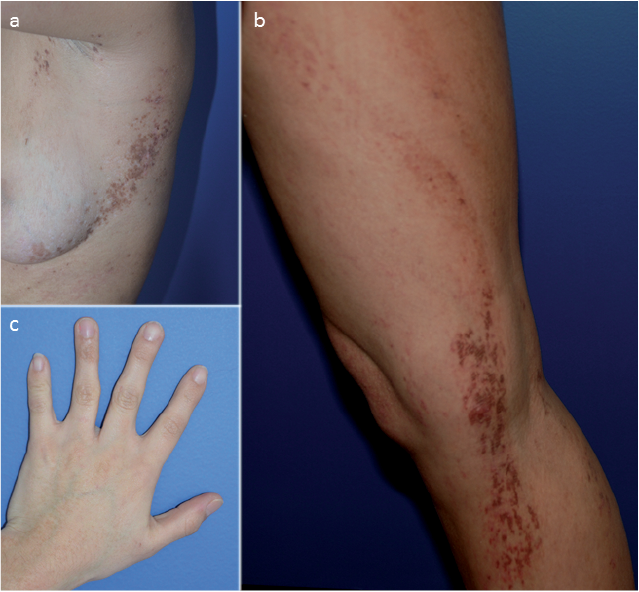
Case 1. A 21-year-old woman presented with skin lesions since birth. She had no medical history and her parents were not consanguineous. At examination, she had linear hyperpigmented and atrophic lesions in a Blaschkoid distribution. Lesions were located on the right arm, right leg and left trunk and involved approximately 10% of the body surface. She also had syndactyly of 2–3 and 4–5 fingers of the left hand (Fig. 1).
Fig. 1. Case 1. (a, b) Linear hyperpigmented and atrophic lesions in a Blaschkoid distribution on the left trunk and right leg. (c) Syndactyly of 2–3 and 4–5 fingers of the left hand.
Histology performed on lesional skin showed some scattered apoptotic cells in the epidermis and pigment incontinence in the superficial dermis. After obtaining written consent, molecular analysis was performed by Sanger sequencing of the coding exons and exon-intron boundaries of the PORCN gene (NM_203475.2, variant D) and multiplex ligation-dependent probe amplification (MLPA) analysis. Both tests were performed on DNA extracted from peripheral blood leukocytes and did not reveal any mutation. Molecular analysis of PORCN was then performed on DNA extracted from saliva and revealed the mutation c.173_178delinsACT (p.Ala58_Gly60delinsAspTrp). This mutation has not been reported previously and was absent from the ExAc control population (10). The percentage of cells carrying the mutation seems low in saliva, but cannot be determined precisely. This mutation is thought to be damaging as it is responsible for the deletion of 3 highly conserved amino acids.
Case 2. A 16-year-old female with healthy, non-consanguineous parents presented since birth with skin lesion in a limited distribution (3% of body surface area). She had Blaschko linear hyperpigmented and atrophic lesions located on the left hemibody: buttock, flank and arm. Examination of the nails revealed longitudinal ridging and V-shaped notches in the distal free margin of the third and fourth nails on the affected hand (Fig. S1). No other associated anomalies were present. Histopathological examination of lesional skin showed a hyperkeratotic epidermis with some maturation disorders (keratinocytes were focally separated by widened intercellular spaces, lost polarity and displayed larger size with pale eosinophilic cytoplasm and large nuclei). After obtaining parental written consent, molecular analysis (Sanger sequencing of the coding exons and exon-intron boundaries of PORCN gene (NM_203475.2, variant D) and MLPA analysis (PORCN-specific synthetic probes and MLPA Reference kit P200kit P200, MRC Holland) were performed on DNA extracted from peripheral leukocytes, affected and unaffected skin. No mutation was detected in peripheral leukocytes or unaffected skin. In lesional skin, we found that the patient was heterozygous for a deletion of the entire PORCN gene c.(?_-182)_(*338_?)del.
We report here 2 new cases of postzygotic mosaicism of FDH in female patients. One mutation was novel. The diagnosis of mosaic FDH was based on the absence of the mutation in peripheral blood-cell tests and a positive result from saliva or affected skin.
The small biopsies taken from lesional skin did not show any dermal hypoplasia, which however is focal and therefore difficult to detect without a normal skin biopsy to compare. Other cases of Goltz syndrome were reported without dermal hypoplasia (11), but with the following histopathological findings: dilated papillary dermal vessels and single or clustered adipocytes in the reticular dermis (12, 13).
The term “genetic mosaicism” is used to describe the coexistence of 2 or more genetically distinct cell populations in one individual (14). It results from sporadic mutations occurring during embryonic development. Only 14 cases of women with mosaic FDH have been reported in the literature (15).
Our 2 patients had a very mild phenotype and limited skin lesions, suggesting that mosaic forms of FDH are associated with milder phenotype, although this is not in accordance with the literature. According to Yoshihashi et al. (16), the clinical presentations of mosaic forms of FDH in women are variable, especially in terms of skin lesions extension and associated anomalies, with mild to severe phenotypes reported. According to Harmsen et al. (17), women with PORCN mutation in a mosaic distribution do not necessarily present with a milder phenotype. They suggest instead that the relative abundance of the mutation in specific developing tissues, such as skin, bone, and eye, is relevant for the development of a full-blown or mild phenotype. Other authors suggest that the extension is linked to an early occurrence of the mutation that can affect both mesodermal germline and ectodermal tissues (18).
Because the clinical manifestations of mosaic FDH can be elusive, in order to avoid false negative results molecular analysis must be performed not only on blood, but also on saliva or, ideally, affected skin (15). In cases of mosaicism, the risk of genetic transmission can vary between 0 % (no mutated germ cells) and 50% (all germ cells mutated).
In conclusion, it is important to be aware of mosaicism for PORCN mutations in FDH, in particular in female patients and independent of the severity of the disease, in order to perform appropriate analyses and deliver accurate genetic counselling.


 Click to show fullsize
Click to show fullsize