


Departments of 1Dermatology and 4Pathology, Hôpital Universitaire Necker-Enfants Malades, 149 rue de Sèvres, FR-75015 Paris, 2Reference Center for Genodermatoses and Rare Skin Diseases (MAGEC), INSERM U1163, Université Paris Descartes – Sorbonne Paris Cité, Institut Imagine, Hôpital Universitaire Necker-Enfants Malades, Paris, France, 3Qatar Biomedical Research Institute, Medical Genetics Center, 5Pathology – Genetics, Sidra Medical and Research Center, Doha, Qatar, 6Yale University School of Medicine, Genetics, New Haven CT, 7University of Iowa, Carver College of Medicine, Iowa City, IA, USA, and 8University of Jordan, School of Medicine, Amman, Jordan. E-mail:smail.hadj@inserm.fr
Accepted Apr 6, 2017; Epub ahead of print Apr 17, 2017
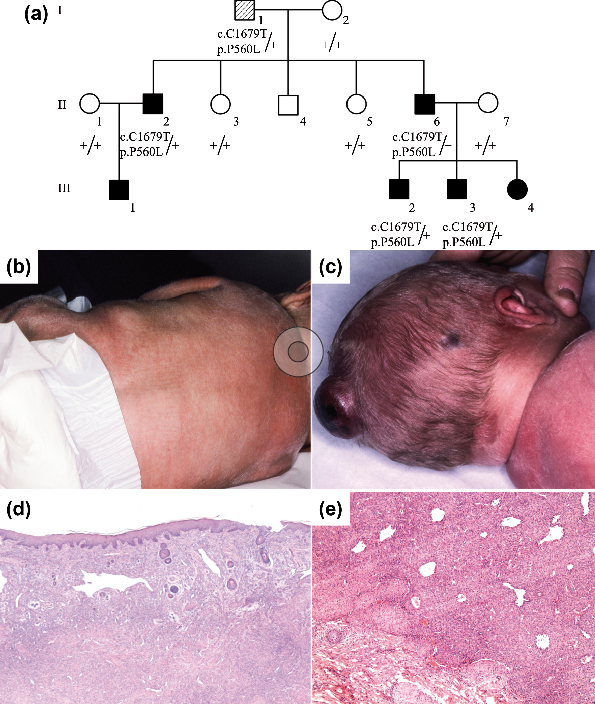
Infantile myofibromatosis (IM; MIM#228550) is a rare disorder of myofibroblastic proliferation and is one of the most common causes of benign fibrous tumour of infancy (1). IM is categorized into solitary, multicentric and generalized forms, with solitary IM being the most common. While solitary IM is characterized by the presence of a single cutaneous nodule, multicentric IM involves the skin, subcutaneous tissues, muscles and bones. Both forms of IM regress spontaneously during childhood. Generalized IM is characterized by visceral involvement, and may have a poor outcome. Although IM is mainly sporadic, 30 families suggestive of autosomal dominant (AD) or autosomal recessive (AR) inheritance have been reported. Recently, heterozygous mutations in PDGFRB and NOTCH3 have been reported in 13 families with AD IM (2, 3). We report here, a 3-generation family with multicentric AD IM and a novel PDGFRB mutation (c.1679C>T; p.P560L).
A male patient (III-2) was referred at birth due to the presence of 4 firm, flesh-coloured subcutaneous nodules; 2 on the scalp, 1 on the left shoulder and 1 on the back (Fig. 1a–c). Histopathology of the dorsal nodule biopsy was consistent with a diagnosis of IM (Fig. 1d, e). Systemic monitoring showed multiple skeletal involvement and a 3-cm nodule of the left psoas muscle, partially infiltrating the spinal canal. No visceral lesions were found. Spontaneous, complete and uncomplicated regression occurred during the first 2 years of life.
Fig. 1. Familial autosomal dominant infantile myofibromatosis (IM): pedigree, clinical and histopathological pictures. (a) Pedigree shows autosomal dominant inheritance. At birth, patient III-2 presented with: (b) 2 firm, flesh-coloured nodules on the back; and (c) 2 firm nodules on the scalp with necrotic evolution. Histopathological analyses of a removed dorsal nodule (patient III-2) showed a lesion located in the dermis and upper subcutis composed of both a spindle-shaped cell proliferation and a vascular proliferation. This angiomatous area, mostly seen at the periphery, is composed of more-or-less dilated capillaries arranged in a haemangiopericytoma-like pattern. The deep dermis is more cellular, less vascular and contains fibrosis. It is likely to be a regressing infantile myofibromatosis lesion (d: haematoxylin and eosin (HE) ×25). The periphery is composed of small myofibroblast fascicles arranged as rounded and hyalinized nodules. Beneath the periphery there are vascular channels, some exhibiting staghorn shapes. Between these the cells are poorly differentiated, polygonal or spindled (e: HE×50).
The patient belongs to a French family of Spanish origin. Six of his relatives presented with AD multicentric IM (Fig. 1a, Table SI). Of note, individual III-3 had a solitary IM subcutaneous nodule 5 years after complete regression of the neonatal lesions. None of the affected relatives presented with skeletal overgrowth, neurological deterioration or dysmorphic features. After obtaining informed consent, DNA extracted from blood leucocytes was analysed by whole exome sequencing for each patient and relative. Re-sequencing of PDGFRB identified a heterozygous missense mutation in exon 12 (c.1679C>T;p.P560L), which segregates with the disease in the family, and identified individual I-1 as the founder despite no history of IM. Variants in NOTCH3 and PTPRG were excluded by whole-exome sequencing.
This case report identified a novel pathogenic PDGFRB mutation in a family presenting with multicentric AD IM. The mutation involves an amino acid that is particularly well-conserved through mammalian species, which segregates with the disease. No other mutation was detected in whole-exome next-generation sequencing.
Familial IM is frequently multicentric, but may exist as solitary or generalized forms in the same family (2, 3). Because most IM lesions are asymptomatic and regress spontaneously, milder expression in relatives may be overlooked, indicating that familial IM is probably more frequent than reported. The mechanism of regression remains unknown.
Heterozygous mutations in PDGFRB have been identified in 13 out the 14 reported families. To date, 4 mutations, including the one identified here, have been reported in IM, the most frequent being c.1681C>T; p.R561C in exon 12 of PDGFRB (2–4). In 4 out of 9 paediatric patients with IM, a second somatic PDGFRB mutation was detected in a removed nodule, suggesting that loss of heterozygosity might explain nodule development, as well as the occurrence of both solitary and multicentric forms in the same family. In addition to PDGFRB, mutations in NOTCH3 and PTPRG have been reported in AD IM (3, 4). Both NOTCH3 and PTPRG belong to the PDGFR signalling pathway (3). PTPRG encodes an enzyme that dephosphorylates PDGFRB, reducing its activity (5). NOTCH3 encodes a transmembrane receptor that directly regulates expression of PDGRB (6). PDGFRB encodes the β-polypeptide of the platelet-derived growth factor receptor, a tyrosine kinase receptor that promotes growth of mesenchymal cells, including fibroblasts and smooth muscle. Beside IM, several other PDGFRB mutations have been ascribed to idiopathic basal ganglia calcification (7), Penttinen syndrome (8), and a distinct overgrowth syndrome with dysmorphic features and neurobehavioral manifestations (9; Table SII). Those mutations in PDGFRB involve the juxta-membrane domain of the protein (2, 3, 9) or its tyrosine kinase domain (2, 3, 8).
While loss-of-function mutation of PDGFRB causes idiopathic basal ganglia calcification, a gain-of-function mutation is responsible for the other 3 disorders. Moreover, PDGFRB activation occurred even in the absence of its ligand. The PDGFRB R561C and N666K mutations involved in AD IM, induced cell proliferation and a strong activation of STAT pathway in the absence of the ligand in vitro (10). Cells transfected with PDGFRB mutant and injected in BALB/c rag2–/– mice promoted tumourigenesis in vivo. The same authors suggested that P584R mutation causing the overgrowth syndrome (9) activates PDGFRβ to a greater extent than R561C mutation. In Penttinen syndrome, the PDGFRB mutations are associated with a ligand-independent hypersensitivity in vitro (8). Thus, distinct gain-of-function mutations occurring in the same gene domain are associated with allelic phenotypes with minimal overlap. Therefore the degree of PDGFRB activation may modulate differently its downstream signalling pathways, explaining the clinical spectrum observed. A modifier gene or a second hit mutation could also be involved.
Mutations in PDGFRA have been associated with several cancers, such as glioblastoma. PDGFRB fusions are reported in myeloid neoplasms (11). Non-specific receptor kinase inhibitors targeting the PDGF receptor, such as imatinib or ponatinib, have shown encouraging results in those diseases (11). Interestingly, p.R561C and p.N666K mutants, involved in IM, are sensitive to imatinib and ponatinib in vitro (10). Further investigations may assess the potential effect of these drugs on aggressive forms of IM.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize