


Assistance Publique-Hôpitaux de Paris, 1Département de Dermatologie, 3Centre National de Référence de l’Histiocytose Langerhansienne, Service de Pneumologie, 4Service de Médecine Interne, 5Département de Pharmacologie Biologique, Hôpital Saint-Louis, Université Paris-Diderot, Sorbonne Paris Cite?; 2INSERM U976, and 6INSERM U1153 CRESS, Equipe de Recherche en Biostatistiques et Epidémiologie Clinique, Paris, France
#These authors contributed equally and should be considered as first authors.
Langerhans cell histiocytosis is a rare histiocytic disorder for which skin involvement and management are poorly described in adults. The aim of this retrospective monocentric study in a national reference centre is to describe the clinical characteristics, quality of life, BRAF mutation status and outcomes of skin involvement in adult patients with Langerhans cell histiocytosis. Twenty-five patients (14 females, mean age 47 years) were included, with a median follow-up of 33 months (range 4–420 months). Patients experienced poor dermatological quality of life despite low body surface involvement. BRAFV600 mutations were detected in 8 of the 18 patients analysed (45%). Eight patients had an associated malignancy. Several treat-ment options were used and consisted of surgery, topical steroids and carmustine, thalidomide, metho-trexate, vinblastine and steroids and cladribine. This study highlights the need to evaluate quality of life and to screen for associated malignancy in adult patients with Langerhans cell histiocytosis.
Key words: Langerhans cell histiocytosis; quality of life; thalidomide; nitrogen mustard; BRAF mutation.
Accepted Apr 18, 2017; Epub ahead of print Apr 19, 2017
Acta Derm Venereol 2017; 97: xx–xx.
Corr: Abdellatif Tazi, Service de Pneumologie, Hôpital Saint-Louis, 1 Avenue Claude Vellefaux, FR-75475, Paris cedex 10, France. E-mail: abdellatif.tazi@aphp.fr
Langerhans cell histiocytosis (LCH) is a rare disorder of unknown aetiology characterized by the infiltration of involved tissues by CD1a/CD207-positive Langerhans-like cells. LCH may involve virtually any tissue/organ and is encountered in patients of all ages, from neonates to the elderly (1–3). LCH cutaneous involvement in children ranges from single self-healing lesions to extensive lesions in life-threatening multisystem disease (4–7). There are few published studies describing LCH cutaneous involvement in adults. Edelbroek et al. (8) described 18 Dutch patients with LCH that first presented in the skin: 5 patients developed a second haematological malignancy (2 acute myelomonocytic leukaemias, 2 lymphomas and 1 histiocytic sarcoma). Another study reported 3 skin-limited LCH adult patients and reviewed 27 other cases in the literature (9). In the large international registry of the Histiocyte Society, describing 274 LCH adult patients (10), skin involvement was present in 37% of patients, almost all with multisystem disease. However, no further information was provided about skin lesions or response to treatment.
Therapeutic options used in adult cutaneous LCH include surgery, radiotherapy, phototherapy, topical or systemic steroids, imiquimod, topical nitrogen mustard, methotrexate, 6-mercaptopurine/azathioprine, vinblastine, thalidomide, and cladribine (11–14). Recent identification of the activating oncogenic BRAFV600 gene mutation in approximately 50% of LCH samples, including the skin, has represented a breakthrough in understanding LCH pathogenesis and offers new therapeutic opportunities with molecules targeting the MAPK pathway (15–19).
This study describes the clinical characteristics, quality of life(QoL), BRAF mutation status and outcomes of LCH with cutaneous involvement in a large cohort of adult patients from a national reference centre.
A retrospective monocentric study was conducted in a national reference centre for LCH. All patients had been included in the French registry for histiocytosis until 2014 and had a diagnosis of LCH with skin involvement according to the Writing Group of the Histiocyte Society criteria (20). Haematoxylin-eosin staining, CD1a, langerin and PS100 immunostaining were performed on all biopsy samples. The study was approved by the Institutional Review Board (CPP Ile de France IV, IRB number 00003835).
Disease state and the response of LCH skin involvement to therapy was clinically assessed for all patients using the International LCH Study Group criteria (21). Patients were categorized as having either non-active skin disease (NAD) (defined as complete resolution) or active skin disease (AD). The response of skin involvement to treatment was classified as better (complete resolution or regression, i.e. incomplete resolution), intermediate (stable or mixed, i.e. new lesions in one site, regression in another site) or worse (progression).
Patients included between 2010 and 2014 (n = 8) had a routine dermatological life quality index evaluation (DLQI). The correlation between DLQI and body surface involvement was performed using the Pearson correlation test (JMP version 11, SAS Institute, USA). Statistical significance was defined as ρ > 0.25 and p < 0.05. Correlations were considered as follows: 0.3 < ρ < 0.5, weak; 0.5 ≤ ρ < 0.7, fair; 0.7 ≤ ρ<0.9, good; and ≥ 0.9, excellent.
Patients included between 2010 and 2014 (n = 8) had a routine dermatological life quality index evaluation (DLQI). The correlation between DLQI and body surface involvement was performed using the Pearson correlation test (JMP version 11, SAS Institute, USA). Statistical significance was defined as ρ > 0.25 and p < 0.05. Correlations were considered as follows: 0.3 < ρ < 0.5, weak; 0.5 ≤ ρ < 0.7, fair; 0.7 ≤ ρ<0.9, good; and ≥ 0.9, excellent.
Sufficient skin tissue material was available in 18 out of 25 patients. DNA was extracted using the QIAamp DNA FFPE Tissue Kit for FFPE tissue (Qiagen, Les Ulis, France), qualified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and quantified using a Qubit® 2.0 fluorometer (Thermo Fisher Scientific, Courtaboeuf, France). BRAF mutation analyses were performed as described previously, using pyrosequencing and high-resolution melting (HRM), on a PyroMark-Q48 Autoprep pyrosequencer (Qiagen, Courtaboeuf, France) and a LightCycler 480 (Roche, Meylan, France), respectively (22).
A total of 25 patients (11 men, 14 women) with a mean age of 47 years (range 21–87 years) were included in the study. Patients’ characteristics and representative clinical pictures are shown in Table I and Figs 1 and 2. Sixty percent of patients had an involvement of intertriginous areas. Scales/crusts and erosions were present in 64% and 40% of patients, respectively. One patient had lichen planus-like nail involvement (onycholysis and striae of the nail bed). Another patient had a subcutaneous histiocytic infiltrate of the thigh and breast. Ten patients (40%) had mucosal involvement (perineal area, 8 patients; mucosal area, 5 patients: erosions, 8 patients; papules/nodules, 2 patients). Lesions were painful in 44% (especially when affecting the scalp) and pruritic in 36% of the patients.
Table I. Characteristics of patients with Langerhans cell histio-cytosis presenting with skin involvement
Fig. 1. Typical skin lesions of Langerhans cell histiocytosis. (A) Crusty papules on the scalp, (B) erythematous papules and macules located on the lower back, and (C, D) erosive papules in (C) submammary and (D) axillary folds.
Fig. 2. Langerhans cell histio-cytosis. (A) Tumorous lesion of the breast. (B) Lichenoid lesions of the nails.
Among the 8 patients tested for DLQI during a LCH skin flare, 5 reported a poor QoL (DLQI >10) despite limited body surface area (BSA) involvement. There was a fair correlation between BSA (median 5%, range 2–11) and DLQI (median 11.5, range 2–21) (ρ = 0.57), although it did not reach statistical significance (p = 0.14).
The histological diagnosis was made on a single biopsy (CD1a-positive, PS100-positive histiocytic dermal infiltrate mostly in the superficial dermis) except in 4 cases, for which 2 biopsies were required (insufficient histiocytic infiltrate on the first biopsy). Other associated histological features included: acanthosis (n = 6), histiocytic epidermotropism (mild, n = 2; moderate, n = 4), histiocytic folliculotropic infiltrate (n = 4), dermal perivascular histiocytic infiltrate (n = 4), neutrophilic infiltrate associated with Langerhans cells accumulation (n = 10), and dermal fibrosis (n = 4). A BRAFV600 mutation was found in 8 of the 18 (45%) patients tested (BRAFV600E mutation, n = 7, BRAFV600K mutation, n = 1).
Eight patients (32%) had skin-limited LCH. Among the remaining 17 patients, other organs that were involved included the lungs (71%), pituitary gland (65%), bones (53%), lymph nodes (35%), spleen and/or liver (29%), ear, nose and throat system (29%) and thyroid (12%). Skin involvement preceded systemic LCH involvement in only 3 patients (by 1 year in 2 cases and 10 years in the other). The median time between LCH diagnosis and the first skin manifestations was 36 months (interquartile range (IQR) 0–163 months). Ten patients experienced skin-limited disease flares despite stable multisystem LCH involvement. Two patients had associated Erdheim–Chester disease (ECD) that occurred simultaneously in one patient and 5 years after LCH diagnosis in the other. Eight patients had an associated malignancy (1 chronic myelomonocytic leukaemia, 1 Hodgkin’s lymphoma, 1 myelodysplastic syndrome, 2 thyroid cancers, 2 breast cancers and 1 prostate cancer) that occurred prior to LCH diagnosis in 6 of these patients.
Median follow-up after diagnosis was 33 months (IQR 12–75 months). Two patients died (one of coronary artery disease, one of unknown cause). Fig. 3 shows the treatments used (at least in 2 patients or more) and clinical outcomes. The median time of evaluation of the response to treatment was 3 months (IQR 3–5 months).
Fig. 3. Treatments used and clinical outcomes of skin involvement in 25 adult patients with Langerhans cell histiocytosis. NAD: non-active disease, AD: active disease.
A watch-and-wait approach was chosen for 2 patients with mild involvement and resulted in stable disease. Twelve patients (48%) were given systemic chemotherapy, which was initiated for systemic LCH involvement in all but one patient. Six of the 7 patients treated with the association of vinblastine and prednisone, and the 5 patients who received cladribine had multisystem LCH involvement. Two patients were given interferon (INF)-α for simultaneous ECD and had a partial response on LCH skin lesions. One of these patients (who had a skin BRAFV600E mutation in both LCH and ECD lesions) was subsequently put on vemurafenib because of inefficacy of IFN-α. Her skin lesions began to improve, but the response could not be evaluated because vemurafenib had to be discontinued after 10 days of treatment (grade 3 drug hypersensitivity reaction).
Various skin-directed therapies were used. Surgery on solitary lesions resulted in a lasting complete response. Topical steroids induced only transient partial responses, whereas topical nitrogen mustard and carmustine were poorly tolerated in 2 of the 6 patients and resulted in complete response in only one case. Thalidomide (50–100 mg/day) induced complete response in 4 of the 6 patients (including 5 patients with perineal and/or oral ulcerations), but was associated with 3 grade 3 adverse events (1 peripheral neuropathy after 14 years of thalidomide use, 1 venous thrombosis possibly linked to thalidomide treatment and 1 grade 3 fatigue). Weekly methotrexate was associated with complete response in 3 patients and progression of skin lesion in the latter. One patient received vinblastine + prednisone for skin involvement and had a complete response. 6-mercaptopurine, pentoxifylline and cotrimoxazole were given in only one patient, and cotrimoxazole resulted in a dramatic complete response in a patient developing disabling vulvar lesions. Acitretin efficacy was interesting in 2 patients. Ten patients needed several different lines of treatment because of a partial response, progression or recurrence of their skin LCH involvement, including 6 patients requiring systemic chemotherapy with cladribine and/or vinblastine. The BRAF mutation status of the patients tested for the presence of BRAFV600 mutations did not correlate with the extent and/or the outcome of the disease.
All patients had LCH chronic skin disease, but some of them had spontaneously regressing lesions even without treatment. These patients ultimately relapsed, however, and no definitive self-healing lesion (formerly known as Hashimoto–Pritzker disease in children) was observed (23). To our knowledge, the effect of cutaneous LCH on QoL has not been evaluated. Among the 8 patients who underwent DLQI evaluation, 5 (63%) had a poor QoL (DLQI >10) although BSA involvement was ≤ 5% in 3 of these 5 patients. This was mostly attributed to painful lesions. The small size of this cohort did not allow us to compare these data with QoL impairment in other skin diseases, such as psoriasis, but the QoL impairment of our patients was quite high given the low BSA involvement (24). These results prompt us to include DLQI evaluation in routine practice for the management of these patients.
Although no specific guidelines exist, skin targeted therapies in adult LCH may include: surgery for isolated lesions, potent topical steroids (first-line), mechlorethamine gel if locally tolerated (currently available for the treatment of cutaneous T-cell lymphoma), thalidomide (25, 26) or weekly low-dose methotrexate (27) and vinblastine + steroids in case of diffuse skin lesions (13). In our study, most patients receiving systemic chemotherapy with vinblastine or cladribine for multisystem LCH improved their skin lesions. More recently, the BRAF inhibitor vemurafenib has been used in selected patients with progressive severe LCH (16, 28–31). One of our patients, who had early drug hypersensitivity syndrome, was treated with vemurafenib. In the literature, it is reported that vemurafenib has been given to 6 patients with skin LCH involvement with a partial or complete skin response (16, 28–31). Adverse events (mainly skin reactions, including squamous cell carcinoma) were common and led to dose reduction or interruption of the treatment. Although promising, the use of BRAF inhibitors should probably be reserved for patients with refractory LCH. A BRAF mutation was found in 45% of our patients. In a series of 315 children with LCH, 172 (54.6%) carried a BRAF mutation, and up to 77% of children with skin involvement had BRAF mutations (32). BRAFV600E has been associated with younger age, and this may explain the lower frequency of this mutation in our adult cohort. The identification of additional mutations in the MAPK pathway in BRAF wild-type LCH lesions suggests that other MAPK-targeted treatments (i.e. MEK inhibitors) might be of use in patients with severe LCH in the future (18).
Eight of our patients had an associated malignancy, including malignant haematological diseases. One patient had chronic myelomonocytic leukaemia and one had myelodysplastic syndrome, which further supports the hypothesis that a clonal abnormality on a common bone marrow precursor may be shared by LCH and myeloid disorders (33). Recently, Johnson et al. (34) reported a patient with a BRAFV600E mutation in ECD skin lesions and a BRAFV600E mutation in a concomitant papillary thyroid carcinoma. The proportion of associated malignancies in our study (32%) was similar to those in a previous series of skin limited LCH (28%) (8), although higher than described previously in larger series of adult patients with LCH (6.2% in (10)).
In conclusion, our study highlights the polymorphic clinical presentation, outcome and response to treatment of LCH in adults. It also emphasizes the need to screen patient’s QoL and associated malignancy. Additional studies are needed to evaluate the use of MAPK inhibitors in severe progressive cases.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize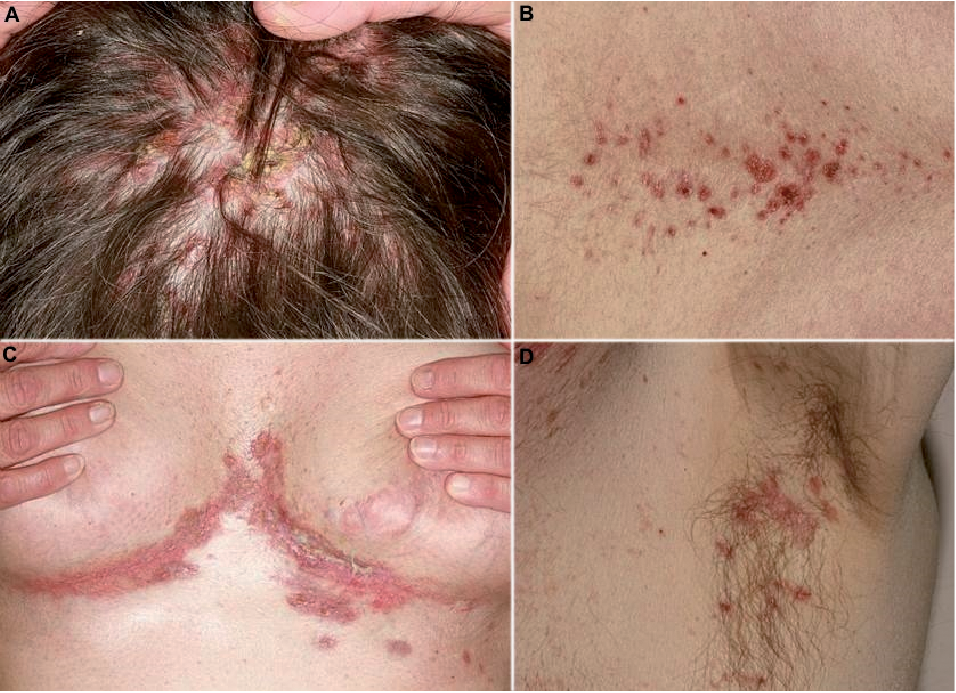 Click to show fullsize
Click to show fullsize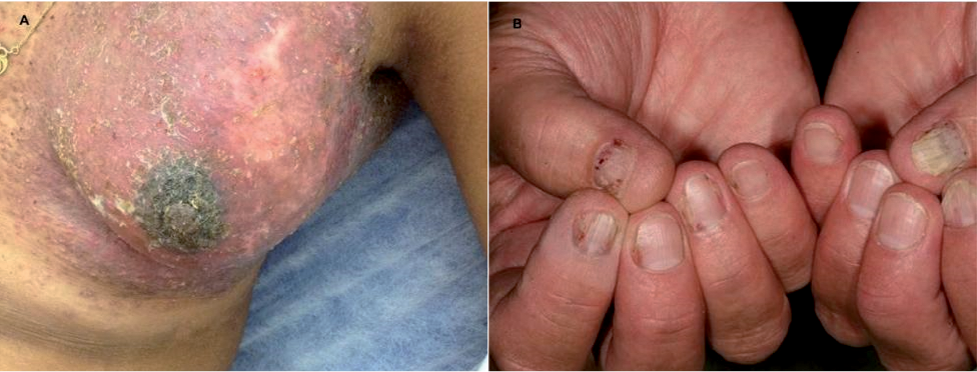 Click to show fullsize
Click to show fullsize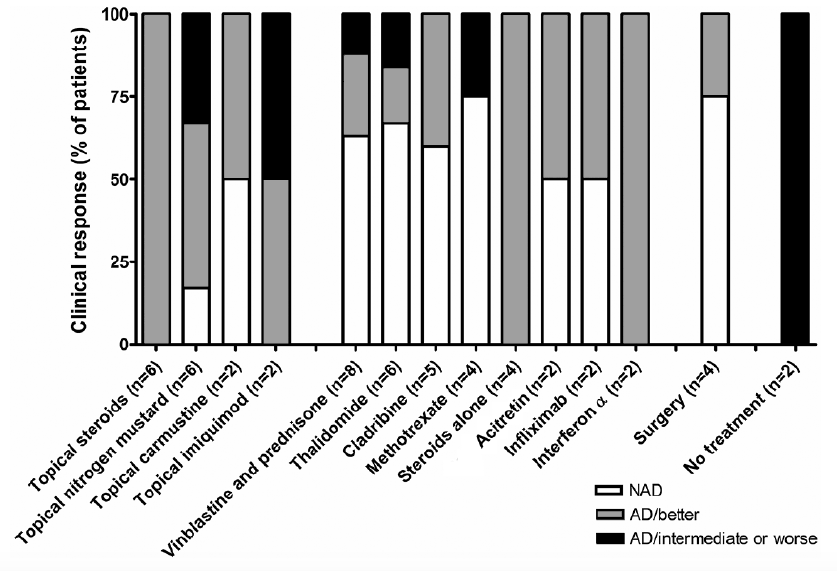 Click to show fullsize
Click to show fullsize