


Department of Dermatology, Tohoku University Graduate School of Medicine, Seriryomachi 1-1, Aoba-ku, Sendai, Miyagi, 980-8574, Japan. *E-mail: saiba@med.tohoku.ac.jp
Accepted Jun 8, 2017; Epub ahead of print Jun 9, 2017
Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (PCAETL) is a rare variant of cutaneous T-cell lymphoma that is characterized by epidermotropic infiltrates of CD8+ cytotoxic T cells, rapid progression, and poor prognosis (1–5). We report here an unusual case presenting with aggressive involvement, including skin, thigh muscle, and intracranial lesions.
A 17-year-old Japanese male, who had been healthy all his life, presented with a 3-month history of swelling of the left parotid gland followed by bilateral upper eyelid swelling with no systemic symptoms. There was no history of allergy, medication or familial medical history. The patient was referred to the Hematology and Rheumatology Department in our hospital for further evaluation. Meanwhile, the patient developed lower extremity myalgia while walking, and weight loss without anorexia. Laboratory examination revealed elevation of serum creatinine phosphokinase (CPK; 956 U/l) and aldolase (31.7 IU/l), no elevation of C-reactive protein, and negative for anti-nuclear antibody, anti-Sm antibody, anti-Jo-1 antibody, anti-PL-7 antibody, anti-PL-12 antibody, anti-EJ antibody, and anti-KS antibody. Serum levels of immunoglobulins IgG, IgA and IgM were within normal range. An electromyogram, magnetic resonance imaging (MRI) of the thigh muscle, and thigh muscle biopsy revealed inflammation in the patient’s thigh muscles.
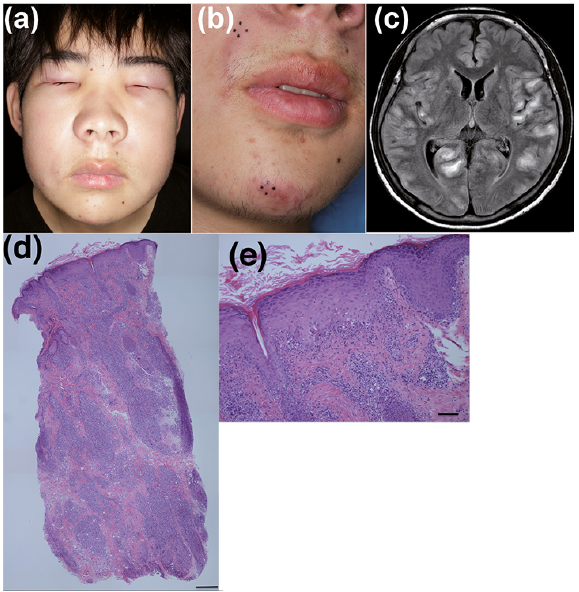
Our department was consulted based on suspicion of dermatomyositis due to those findings. Physical examination revealed facial swelling, especially on the bilateral upper and lower eyelids with some erythema, suggestive of heliotrope eyelids (Fig. 1a). Gottron’s sign or nail-fold capillary changes were not observed on the patient’s hands. The first skin biopsy of the left eyelid revealed vacuolar degeneration accompanied by lymphocyte infiltration into the dermo-epidermal junction and the follicular infundibulum. Since there were several atypical cells among the infiltrating lymphocytes, a conclusive diagnosis of dermatomyositis was suspended because we could not exclude the possibility of cutaneous lymphoma. At this time, the soluble interleukin-2 receptor level was elevated to 3,067 U/ml. Oral corticosteroid, 50 mg/day, had been administered to the patient for treatment, which was effective for the facial and eyelid swelling. However, the resolution of diffuse swelling revealed asymptomatic erythematous induration on the nasolabial fold and the chin (Fig. 1b). A skin biopsy was taken from the lesion. One day after the biopsy the patient suddenly developed systemic fatigue, headache, anorexia, nausea, difficulty moving the left arm, signs of meningeal irritation and cerebellar ataxia. Magnetic resonance imaging (MRI) of the brain demonstrated an increased fluid-attenuated inversion recovery (FLAIR) signal deep in multiple parts of the cerebral cortex (Fig. 1c), suggesting posterior reversible encephalopathy syndrome, although no abnormal signal had been found in the brain MRI that had been taken approximately 2 weeks previously. Electroencephalogram and cerebral scintigraphy revealed severe brain damage, especially in the right hemisphere. Whole-body screening with (18)F-fluorodeoxyglucose positron emission tomography/computed tomography showed no significant abnormalities. The skin biopsy from the lesion revealed massive infiltration of atypical lymphocytes into the dermo–epidermal junction and epidermis, with their dermal infiltration constituting node-like structures in the dermis and subcutaneous infiltration (Fig. 1d, e). Immunohistochemical staining showed that these atypical cells were: CD45+, CD2+, CD3+, CD5–, CD7 weak+, CD4–, CD8 weak+, CD56–, CD30–, CD68–, TIA-1+, granzyme B+ and 60–70% positive for Ki-67. A cerebral biopsy from the cortex demonstrated perivascular infiltration of atypical lymphocytes that surrounded vessels with swollen endothelial cells. Infiltration of atypical lymphocytes was also detected in muscle in a re-evaluation of the muscle biopsy. Based on these clinical features, together with MRI and histological findings, the patient was finally diagnosed with PCAETL involving the thigh, muscle and brain. The patient was initially treated with high-dose dexamethasone to reduce brain damage, followed by chemotherapy consisting of etoposide, cyclophosphamide, vincristine, doxorubicin, and prednisolone (EPOCH regimen), which was, unfortunately, ineffective. Allogeneic cord blood transplantation finally prevented disease progression and improved the intracranial lesions in brain MRI. After tranplantation the lymphoma progression was halted, so that the patient became able to move after rehabilitation.
Fig. 1. Clinical and dermatopathologic features. (a) Facial swelling, especially on the bilateral upper and lower eyelids with erythema, on the first visit. (b) Erythematous induration on the skin of the nasolabial fold and the chin during treatment with oral corticosteroids. (c) A magnetic resonance imaging (MRI) study of the brain revealed an increased fluid-attenuated inversion recovery (FLAIR) signal in multiple parts of the cerebral cortex. (d) Skin biopsy of an erythematous induration revealed dense inflammatory infiltration from the dermo-epidermal junction to subcutaneous tissue, and (e) vacuolar degeneration and epidermotropic infiltration of atypical lymphocytes (haematoxylin-eosin staining. Low magnification ×40, scale bar: 5 µm. High magnification ×100, scale bar: 500 µm.).
PCAETL, which was first described by Berti et al. in 1999 (1), is an extremely rare variant (less than 1% of primary cutaneous lymphomas) and still a provisional entity of peripheral T-cell lymphoma not otherwise specified in the WHO/EORTC classification of cutaneous lym-phomas (1–5). The clinical course is aggressive, with a median survival of 12–32 months and a 5-year survival of 18–32% reported in previous studies (1–3). It is known that the lesion involves unusual sites, including the testes, lung, oral cavity and central nervous system (CNS), but not the lymph nodes. Histologically, this lymphoma shows a prominent epidermotropism of atypical lympho-cytes with a CD8+ cytotoxic phenotype (1).
Clinical features are characterized by rapid onset of patches, plaques, eruptive papules, nodules, pigmented macules and tumours, accompanied by ulceration or necrosis (1, 6). Our case initially presented with swelling of the face, especially of the upper and lower eyelids with erythema, and thigh muscle pain while walking. The laboratory data showed elevation of CPK and aldolase. These findings are similar to those of dermatomyositis, which is an autoimmune disease that involves skin and muscles. However, there has been no report of PCAETL with dermatomyositis-like clinical manifestations.
PCAETL has been reported to cause CNS disorder (1). Unfortunately, the present case also suddenly developed hemiplegia, meningitis, and cerebellar ataxia approximately 4 months after the appearance of the earliest cutaneous symptoms. Two weeks before the patient developed CNS symptoms, an MRI of the brain had revealed no abnormal signal in the intracranial lesions, suggesting that the disease had rapidly progressed even within these 2 weeks. Berti et al. (1) reported that 3 out of 17 patients (17.4%) with PCAETL had CNS involvement. This percentage is extremely high compared with the 1.3–1.5% of patients with mycosis fungoides (MF) and the 8.5–10% of patients with peripheral T-cell lymphoma who have CNS involvement (7). To date, the reason why PCAETL preferentially infiltrates the CNS is unknown. Perhaps some adhesion molecules expressed by CD8+ T cells might make it possible to disrupt the blood–brain barrier and invade brain tissue.
The differential diagnoses were PCAETL, CD8+ variant of MF, subcutaneous panniculitis-type T-cell lym-phoma (SPTL), gamma-delta T-cell lymphoma, natural killer/T-cell lymphoma of nasal type, blastoid precursor T-cell lymphoma, and lymphomatoid papulosis type D. SPTL was excluded because the infiltration of atypical lymphocytes along the dermo-epidermal junction was so conspicuous and we could not detect the rimming of atypical lymphocytes around adipocytes, which was one of the pathognomonic signs of SPTL. Other diseases were ruled out by the clinical features, aggressive clinical course and histological and immunohistochemical findings of this case. To differentiate CD8+ variant of MF, we used the criteria presented by Nofal et al. (8): rapid onset, cutaneous nodules from the beginning, epidermotropism of CD8+ T cells along the basal epidermal layer, infiltration deep into subcutaneous fat, folliculotropic infiltration, and CNS involvement.
Treatment options include radiation, chemotherapy, or blood cell transplantation. Liu et al. reported a case that had obtained a complete remission with allogeneic bone marrow transplantation (9). In our case, allogeneic cord blood transplantation with a conditioning regimen consisting of cyclophosphamide, cytarabine, and total-body irradiation prevented disease progression.


 Click to show fullsize
Click to show fullsize