


1Faculty of Health Sciences, University of Southern Denmark, 2Department of Pathology, Odense University Hospital, and 3Department of Dermatology and Allergy Centre, Odense University Hospital, Odense, Denmark
Henoch-Schönlein purpura is the most common childhood vasculitis, but may also affect adults. This article reviews the literature since 2011 on advances in diagnosis, clinical disease manifestations, pathophysiology and treatment of Henoch-Schönlein purpura. The clinical manifestations are thought to arise from IgA depositions in blood vessel walls in the affected organs, mostly skin, gastrointestinal tract, joints and kidneys. Corticosteroids may be effective in rapid resolution of renal manifestations and treating joint and abdominal pain, but they are not proven effective for treating organ manifestations and complications, such as glomerulonephritis, bowel infarction or intussusception. Mycophenolate mofetil or cyclosporine A may be better treatment choices in case of renal involvement. Other immunosuppressive and immunomodulating drugs, such as rituximab and dapsone, are promising, but larger studies are needed to confirm these findings. Cancer screening should be considered in older males diagnosed with Henoch-Schönlein purpura.
Key words: Henoch-Schönlein purpura; vasculitis; immunoglobulin A; corticosteroids.
Accepted Jun 22, 2017; Epub ahead of print Jun 27, 2017
Acta Derm Venereol 2017; 97: xx–xx.
Corr: Liv Eline Hetland, Faculty of Health Sciences, University of Southern Denmark, Henriettevej 36, DK-5000 Odense, Denmark. E-mail: livelinehetland@gmail.com
Henoch-Schönlein purpura (HSP) is the most common childhood vasculitis, affecting 10–20 children per 100,000 per year. More than 90% of patients are under 10 years of age, with a mean age of 6 years (1, 2). HSP is a leukocytoclastic vasculitis involving small vessels (3). Its clinical presentation includes cutaneous palpable purpura, joint pain, renal involvement, colicky abdominal pain and gastrointestinal bleeding. Most cases of HSP occur in autumn and winter. Proposed triggers include upper respiratory tract infections, medications, vaccinations, and malignancies (4, 5). The pathophysiology behind HSP is not yet completely understood. HSP is generally self-limiting and harmless, but concomitant nephritis may cause severe complications. The proportion of patients having renal involvement varies between 20% and 80% in the literature (6). The estimated incidence of nephrotic or nephritic syndrome is ~7% of all HSP cases, and 1% of patients develop end-stage renal failure (7, 8). HSP nephritis (HSPN) usually occurs within 1–2 months after the onset of HSP.
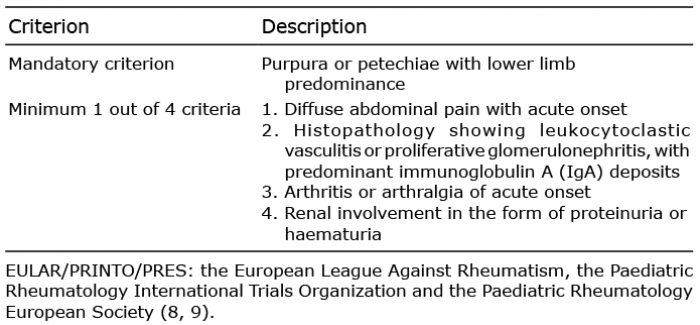
The diagnosis of HSP is criteria-based. The European League Against Rheumatism (EULAR), the Paediatric Rheumatology International Trials Organization (PRINTO) and the Paediatric Rheumatology European Society (PRES) published a revised set of criteria in 2010, with high sensitivity and specificity (9).
The severity and organ involvement of the disease dictates the treatment. In general, treatment for HSP without renal involvement is symptomatic. HSPN is commonly treated with corticosteroids or other immunosuppressive and modulating drugs. Existing studies are inconclusive regarding drug of choice.
A systematic search of the literature was performed in PubMed and Embase databases. The MeSH term in the PubMed database was “Henoch Schönlein purpura”, limited to the title, for articles published between 2011 and 2016. This search yielded 508 articles. In the Embase database, the keywords were “henoch”, “henoch schönlein purpura”, “purpura” and “schönlein”. The same limitations applied. This search yielded 1,503 articles. The number of articles chosen for further reading was 300. The references in the articles selected for this review were investigated further.
HSP diagnosis is based on clinical criteria. The revised criteria developed by EULAR/PRINTO/PRES were published in 2010, and are the gold standard for the diagnosis of HSP (Table I). The sensitivity is 100% and specificity 87%, when applied to children (9). One study reviewed these criteria to assess applicability to adults, and found a diagnostic sensitivity of 99.2% and specificity of 86%, supporting its use for all patients with HSP (10). There are currently no specific biomarkers useful for diagnosis of HSP. Some biomarkers can show activity and prognosis of the disease, but none have proven clinically useful (11–13).
Table I. Diagnostic criteria for Henoch-Schönlein purpura (HSP), as developed by EULAR/PRINTO/PRES
Skin biopsies are the gold standard for diagnosing any cutaneous vasculitis. IgA-predominant vascular deposits are characteristic for HSP, but not sufficient for the diagnosis of HSP, as these deposits can be found in other vasculitic syndromes, erythema nodosum and venous stasis-related conditions (14).
The histological features of HSP involving the skin are those of a leukocytoclastic vasculitis primarily affecting the small superficial vessels. The vessel walls are infiltrated by neutrophil granulocytes, which partly degenerate and form nuclear dust (leukocytoclasia), located amongst extravasated erythrocytes (purpura) in the surrounding dermis. The vessel walls are thickened and might be necrotic due to exudation of neutrophils and variable amounts of fibrin. On direct immunofluorescence IgA and, eventually, complement C3 can be seen deposited in the vessel walls (Fig. 1). The process is dynamic and not all of these features might be seen in a single biopsy. Positive histology is one of the non-mandatory criteria in the diagnostic criteria developed by EULAR/PRINTO/PRES (9). Several authors argue that skin biopsies are not indicated unless the diagnostic criteria based on clinical presentation are not met, or if the presentation is atypical or incomplete (15–17). An abstract included in the annual American College of Rheumatology/Association of Rheumatology Health Professionals (ACR/ARHP) meeting in 2016, describes leukocytoclastic vasculitis in 205 of 216 (92%) adult patients with HSP (18). Direct immunofluorescence revealed IgA deposition in dermal blood vessels in 174/216 (81%) patients. One study analysed patients diagnosed with HSP and concomitant histological diagnosis of cutaneous leukocytoclastic vasculitis. They found IgA positivity associated with HSP with a sensitivity of 81% and a specificity of 83% (19).
Fig. 1. (A) Punch biopsy from skin showing neutrophils accentuated around and in superficial vessel walls. There are extravagated erythrocytes, leukocytoclasis and focal exudation of fibrin (haematoxylin-eosin stain x 150). (B) Positive IgA immunofluorescence (x 200) in a patient with Henoch-Schönlein vasculitis.
The classic tetrad of HSP includes palpable purpura, joint pain, gastrointestinal complaints, and renal involvement (Fig. 2). These clinical manifestations may develop over the course of days to weeks. The order of presentation may vary. The initial presentation is usually with purpura and joint pain (20). In a review of 150 children with HSP, all patients had palpable purpura (21). Seventy-four percent of patients had joint involvement. Renal and gastrointestinal involvement was seen in 54% and 51% of patients, respectively. A survey from 2016 analysed clinical symptoms in 260 adults with HSP (18). At diagnosis, 100% of patients presented with purpura. Joint involvement, glomerulonephritis and gastrointestinal involvement were found in 62%, 70% and 53% of patients, respectively. One study compared symptoms in 75 adults vs. 208 children with HSP (22). Children had joint involvement and abdominal pain more often than adults. There were more adult cases with lower extremity oedema and hypertension. Less common clinical manifestations of HSP include cerebral vasculitis, testicular haemorrhage and interstitial pulmonary haemorrhage (23).
Fig. 2. (A and B) Classical skin lesions of Henoch-Schönlein purpura, with palpable purpura on the extremities. (C) Arthritis and purpura on the lower extremity. (D) Bullous and necrotic lesions of the lower extremities in a patient with complicated Henoch-Schönlein purpura.
The rash often begins with petechiae and palpable purpura. Occasionally, erythematous macular or urticarial wheals may also appear. The lesions may merge and evolve into ecchymoses, petechiae and palpable purpura, and could also turn into bullous or necrotic lesions (8, 20). Gravity-dependent areas and pressure points may favour the localization of rashes. The rash is especially common on the lower extremities and buttocks. The reason for this phenomenon is unclear, but some researchers have suggested that gravity causes immune complexes to deposit and incite inflammation in dependent areas (24). Up to one-third of patients experience trunk and upper extremity involvement (25). When the haemorrhagic skin lesions disappear, hemosiderin deposits will discolour the skin for weeks (26).
Fifteen percent of patients with HSP present arthritis as the initial symptom (21). Temporary, non-destructive poly-arthralgias involving the knees and ankles are usually seen. Hands and feet may also be affected. The affected joints are painful, swollen and have reduced function (27). Arthralgia or arthritis involving only a few joints occurs in approximately 75% of children with HSP (23).
In 10–40% of patients, gastrointestinal manifestations may precede the onset of skin purpura (11). The main explanation of these symptoms is immune complex deposition in the intestinal vessel walls. A prospective trial performed by Jauhola et al. (27) included 221 patients with HSP less than 16 years of age. Abdominal pain was found in 57% of these patients. Melaena and haematemesis were present in 18 and 2 of these patients, respectively. The article emphasized measurement of serum albumin levels in all patients with HSP, as hypoalbuminaemia in the absence of proteinuria can indicate intestinal involvement and protein loss, also in patients without abdominal symptoms. A recent study on children showed that faecal calprotectin might also be a reliable marker for gastrointestinal involvement in HSP (28). In severe cases, gastrointestinal symptoms may mimic an acute surgical abdomen. Complications of abdominal involvement include perforations, intussusception and bowel infarctions. This can lead to death if surgical intervention is not initiated in time (29).
In 20–55% of children with HSP, renal symptoms usually follow the onset of rash within 1–3 months (23). HSPN develops when the renal parenchyma is affected and HSPN is the leading cause of morbidity from this disease (30). Manifestations range from microscopic haematuria and mild proteinuria to nephrotic and nephritic syndrome and renal failure. Hypertension may develop at the onset or during recovery of HSP. The most common finding is isolated microscopic haematuria that usually develops within 4 weeks after onset of the disease. Most HSPN cases are mild, and the chances of recovery are good (8). Children with no renal symptoms during the first 6 months after the onset of HSP are not likely to develop long-term renal damage (31).
Although rare, HSP can have neurological manifestations. Clinical presentation appears from 2–4 weeks into the course of HSP (32). Most frequent symptoms are headaches, seizures and more non-specific changes in the central nervous system (CNS), which entail emotional instability, irritability, dizziness and behavioural changes. Other rare complications include ataxia, intracranial haemorrhage, mononeuropathy, and acute motor sensory axonal neuropathy.
Upper respiratory tract infections precede a majority of HSP cases and multiple case studies propose a correlation between practically all respiratory pathogens and HSP. Streptococcus strains and Parainfluenza virus are the most commonly associated pathogens, and in children Human Parvovirus B19 is a frequent viral trigger (23, 33, 34). The interaction between leukocytes and vascular endothelial cells contributes to the pathogenesis of HSP. Endothelial damage, perivascular leukocytic infiltrates, chemokines and cytokines are important factors in this process (35, 36). Vascular deposition of IgA1-containing immune complexes plays a pathogenic role (37). Complement activation, cellular damaging and IgA deposition suggest that HSP is an IgA-mediated dysregulated immune response to an antigen (4). Through binding and activation of complement factors, IgA cross-reacts with endothelial cells and damages the cells. Advances in technology have allowed updated data on the function of the human immune system and what happens when it fails. In HSP, the dysregulated immune response may result in inflammation and vasculitis without a granulomatous reaction (26). Several antibodies, cytokines, chemokines, receptors, and transmembrane proteins have been found to be involved. Amongst these are cytokines, such as tumour necrosis factor alpha (TNF-alpha), interleukin (IL)-6, and IL-8 (38). Studies showed that Toll-like receptors TLR-2 and TLR-4 were upregulated in children with HSP. These proteins are mainly expressed, regulated and produced by cells of the immune system, including macrophages and lymphocytes. They may also arise from non-immune cells, such as epidermal cells, fibroblasts, kidney podocytes and mesangial cells (39, 40). One study stated that plasma levels of IgA anti-beta2-glycoprotein I antibodies are increased in childhood HSP (41). They are thought to have a strong association with heavy proteinuria and joint manifestations.
Genetic predisposition may contribute to the development of HSP. An Israeli study demonstrated that 10% of patients with HSP were homozygous for mutations of the gene encoding MEFV (the gene defective in familial Mediterranean fever), and additionally 17% had heterozygous defects (42). In comparison, in a randomly selected cohort in the general Israeli population, only 1–2% carried 2 mutant alleles. MEFV encodes the protein pyrin/marenostrin, which regulates caspase-1-activation and IL-1B production. Human leukocyte antigen haplotypes may also play a role in susceptibility to HSP. A study on children with HSP showed an increased risk for the development of HSP in children carrying human leukocyte antigen A2, A11 and B35 antigens, and a reduced risk in the carriers of HLA A1, B49 and B50 antigens (43).
The treatment strategies for HSP remain controversial. The general agreement is to base therapy on the presence or absence of renal involvement. Without renal involvement, the treatment is purely symptomatic. Pain medication, rehydration therapy and surgery for intussusception are examples of this. In case of skin necrosis with ulceration, wound therapy may also be necessary. Compression therapy may be used when oedema of the lower legs occurs. There is still no consensus on treatment of HSP nephritis and other severe complications.
There is disagreement between researchers and clinicians whether to use corticosteroids. Over the last 6 years there have been some studies that can help clarify the applicability of corticosteroids. In 2013, Dudley et al. (44) conducted a double-blinded, randomized, placebo-controlled trial of corticosteroids in 352 children with recent-onset HSP and no or minor renal involvement. Prednisolone was administered for 2 weeks, and the conclusion was, that treatment with corticosteroids demonstrated no benefit over placebo in reducing the risk of proteinuria 12 months after the onset of HSP. In addition, the trial did not determine how patients with more severe renal involvement during the course of the disease should be treated. Earlier studies, performed in 2004 and 2006, including an 8-year follow-up, showed the same results, with no long-term benefit (45, 46). Renal manifestations, such as haematuria and proteinuria, were not prevented after 28 days of corticosteroid treatment, but were resolved faster compared with patients receiving placebo. At 6-month follow-up, 61% of patients had resolved renal manifestations compared with 34% of placebo patients. This study showed the greatest efficacy in patients over 6 years of age presenting with mild renal manifestations at inclusion, and suggests the potential use of corticosteroids in mild cases in order to alter the course of renal involvement. The studies also found statistically significant results regarding treatment of extra-renal symptoms. Abdominal and joint pain were reported less frequently in patients receiving corticosteroid treatment vs. placebo. There was no difference between the groups with regard to skin manifestations.
An updated Cochrane review from 2015 aimed to clarify the different treatment options for kidney involvement in patients with HSP, compared with placebo or other treatments (47). Five randomized controlled trials formed the basis for this review, and none of the studies presented evidence for the benefit of corticosteroid treatment for renal involvement.
A randomized controlled trial compared methylprednisolone and cyclosporine A as treatments for HSPN (45). Twenty-four children with nephrotic-range proteinuria or crescentic HSPN in kidney biopsies were included. Eleven patients were treated with cyclosporine A, and all achieved resolution of proteinuria within 3 months. Of the 13 patients receiving methylprednisolone, 6 did not achieve resolution of proteinuria, and were treated with cyclosporine A as an alternative. Five of these patients responded to cyclosporine A. Biopsy outcomes after 2 years were the same in the 2 treatment groups, but cyclosporine A showed faster regression of proteinuria and a higher frequency of response.
In one study, 12 children with steroid-resistant nephrotic-range proteinuria received mycophenolate mofetil and all responded to the treatment with no relapses occurring (48). One systematic review of 10 RCTs involving 426 patients aimed to assess the safety and efficacy of myco-phenolate mofetil for HSPN vs. other immunosuppressive therapies (49). The conclusion was that the efficacy of mycophenolate mofetil was better than cyclophosphamide after 12 months. Cyclophosphamide and prednisone caused more side-effects than mycophenolate mofetil.
Dapsone is a drug known for its anti-inflammatory and immunomodulatory effects. It has been prescribed in a few individual cases. One patient presented chronic skin lesions of the legs, which disappeared within 24 h after initiating dapsone, and the patient remained asymptomatic with a lower dose as maintenance therapy (50). In 3 other cases, dapsone was initiated as treatment for chronic, recurrent, persisting purpuric skin lesions. The outcome was complete healing in all cases (51).
Rituximab is an anti-CD20 antibody, functioning as a B-cell inhibitor. The efficacy of this drug on HSP has been observed in several case studies over recent years. One case report described a patient with severe skin lesions and moderate kidney involvement, who after 2 doses of rituximab showed complete remission (52). Another case study presented a patient with relapsing HSP, who was unresponsive to corticosteroid treatment and only mildly responsive to cyclophosphamide, who after 5 courses of rituximab experienced complete remission of HSP (53). A patient with end-stage renal disease and corticosteroid-dependent HSP likewise had complete remission after 2 infusions of rituximab (54).
A Cochrane Review regarding treatment of kidney disease in HSP assessed 2 studies in which the efficacy of cyclophosphamide was evaluated (47). One of the studies included 56 children with significant HSP-associated kidney disease who received either cyclophosphamide or supportive treatment (55). There was no significant difference in the risk of persistent kidney disease of any severity during follow-up between the 2 treatment groups. The other study compared cyclophosphamide plus corticosteroids vs. corticosteroid monotherapy in 54 adults with severe HSP. Adding cyclophosphamide provided no benefit compared with corticosteroids alone.
Anticoagulants, such as warfarin, dipyridamole and acetylsalicylic acid (ASA), have been used alongside immunosuppressive agents, supported by the possible role of fibrin deposition in glomerular crescent formation (40). One study proposed heparin as prevention for HSP-related kidney disease (47). The use of anticoagulants is generally not well documented and may cause serious side-effects. As such, their use is not justified. Plasmapheresis has been proposed to remove circulating IgA1 and IgA1-complexes, which are responsible for organ manifestations. Several case reports relate the dramatic improvement of extra-renal symptoms after plasma exchange (40). However, the general consensus about plasmapheresis is, that it seems effective as an adjuvant therapy in a multi-faceted treatment regimen, but larger randomized controlled trials are needed to conclude this (56). Tonsillectomy has been proposed as a treatment and prophylaxis based on several case reports, as HSP is often triggered by an upper respiratory tract infection (57). However, a suggested link between chronic tonsillitis and HSP has not yet been proven.
Vasculitis is associated with cancer with an incidence of ~2–5%, and the majority of cases are related to haematological malignancies. The onset of vasculitis may appear before, during or after the cancer diagnosis (5, 58). HSP is more commonly associated with solid tumours than with haematological malignancies. The gastrointestinal tract, respiratory organs and urinary tract are the most affected organs (29, 58–60). These patients are mostly male, approximately 60 years of age, and screening for cancer in this subgroup could be indicated in the case of unexplained development of HSP, especially if the rash spreads to the trunk and upper extremities (58, 60).
The presentation and diagnostic criteria of HSP are well described in the literature. In recent years, there has been great progress in research, leading to a better, but still not complete, understanding of the pathogenesis. The majority of cases are preceded by an upper respiratory infection, and patients show vascular depositions of IgA immune complexes in several organ systems, which lead to the disease manifestations. Screening for cancer should be considered in adult patients, especially in males approximately 60 years of age who have HSP with no preceding infection,. Treatment includes symptomatic, and eventually immunosuppressive and immunomodulating, agents, e.g. mycophenolate mofetil or cyclosporine A in the case of renal involvement. We recommend that immunosuppressants and immunomodulators are restricted to chronic, persistent, recurrent or complicated cases. Future multicentre studies in children and adults should determine whether corticosteroids are indicated, and assess other possible steroid-sparing drugs, such as rituximab or dapsone in individual organ manifestations. From a clinical point of view, an evidence-based treatment algorithm for HSP based on disease manifestations is needed.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize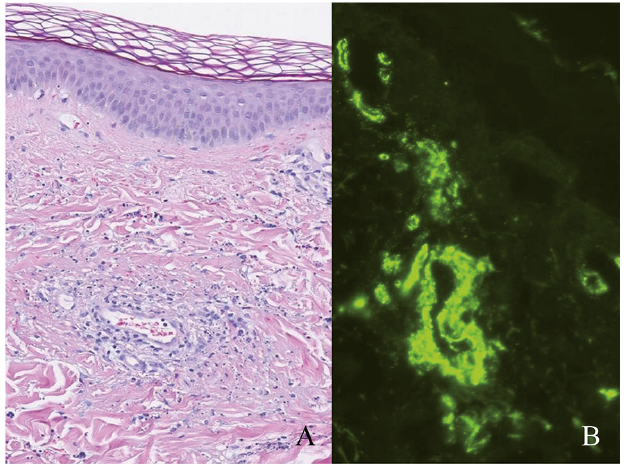 Click to show fullsize
Click to show fullsize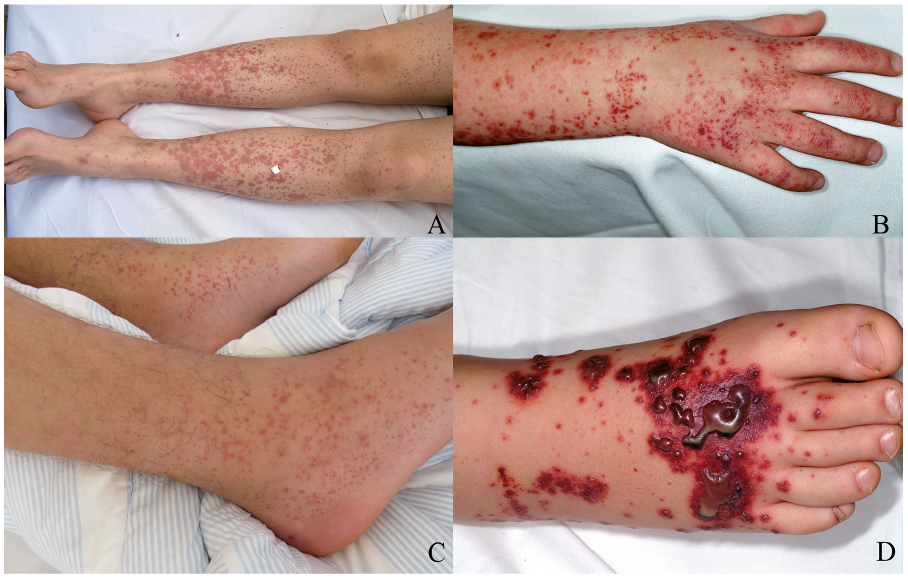 Click to show fullsize
Click to show fullsize