


Departments of 1Immunology, 2Dermatology, and 3Pathology, Hôpital Saint-Louis, Assistance Publique-Hôpitaux, INSERM UMRS_1165, Université Paris Diderot, 1 avenue Claude Vellefaux, FR-75010 Paris, France. E-mail: marion.malphettes@aphp.fr
Accepted Aug 23, 2017; Epub ahead of print Aug 23, 2017
Some dermatological entities are strongly associated with the presence of monoclonal gammopathy (MG) and should be referred to as monoclonal gammopathy of cutaneous significance (MGCS), as proposed recently (1). Acquired bullous dermatosis is a rare complication in this setting. Most reported cases of bullous dermatosis complicating MG are related to systemic light chain (AL) amyloidis (2). We report here an acquired bullous dermatosis mimicking bullous pemphigoid (BP) presenting as the first manifestation of systemic light chain deposition disease (LCDD).
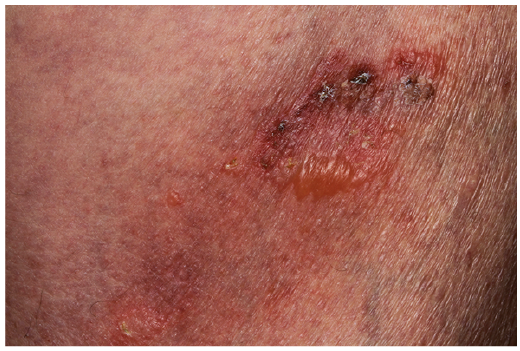
A 77-year-old man was referred to our institution in 2012 for a pruriginous erythematous bullous eruption of the trunk and scalp (Fig. 1). He had multiple, tense, irregular-shaped bullae, either on erythematous skin or in place of recent scars, together with excoriations. There was no Nikolsky’s sign, and no mucosal involvement. Skin biopsy showed a subepidermal blister associated with an infiltrate of neutrophils and eosinophils, consistent with autoimmune bullous dermatosis. Direct immunofluorescence (DIF) with anti-IgG heavy chain, anti-IgA heavy chain, anti-IgM heavy chain and anti-C3 antibodies was negative. Indirect immuno-fluorescence (IF) on salt-split skin was negative. Serum anti-BP 230 antibodies were slightly positive, while anti-BP180 and anti-desmoglein 1 and 3 antibodies were negative. A diagnosis of atypical BP was suggested. Topical corticosteroids were started and were initially effective, but the cutaneous disease subsequently evolved in a relapsing/remitting manner.
Fig. 1. Left thigh, showing tense, irregular-shaped bullae on erythematous skin together with excoriations.
In 2014, flare-up occurred when attempts were made to taper the dosage of topical corticosteroids. A new skin biopsy revealed a subepidermal blister associated with a predominantly lymphocytic moderate infiltrate and rare neutrophils or eosinophils (Fig. S1). At this time, skin DIF showed no IgG, IgA or IgM heavy chains, nor C3 deposits. Immunoelectrophoresis performed in November 2014 demonstrated a monoclonal IgA lambda paraprotein, and the patient was referred to the haematology outpatient clinic. At the clinic in February 2015, he reported gradual onset of dyspnoea and lower limb oedema, progressing for 3 months. Physical examination revealed symmetrical peripheral oedemas of the inferior limbs. There was no jugular venous distension or lung crackles. Skin examination revealed numerous haemorrhagic tense blisters spread over the trunk and limbs. The rest of the systemic examination was unremarkable.
Laboratory tests revealed a markedly elevated concentration of urine protein (12 g/24 h), consisting largely of albumin, with traces of lambda free light chains (FLC). Serum albumin level was 28.9 g/l. Limb oedema was attributed to nephrotic syndrome. Other laboratory tests revealed kidney function to be within the normal range. Immunofixation confirmed the presence of a monoclonal paraprotein IgA-lambda in the serum (0.7 g/l). Serum lambda FLC were increased, at 1,215 mg/l (normal 5.7–26.3 mg/l), while kappa FLC were normal at 11.4 mg/l (normal 3.3–19.4 mg/l). Bone marrow aspirate showed 10% atypical plasma cells. Skeletal X-ray did not detect any lytic bone lesion.
Another skin biopsy showed a sub-epidermal blister associated with a slight inflammatory lymphocytic and neutrophilic infiltrate. No amyloid deposits were seen on Congo red staining. Epidermal and vascular basement membranes were slightly thickened. Skin DIF revealed abundant granular lambda light chain (LC) deposits at the dermoepidermal junction and, to a lesser degree, around dermal vessels, with no kappa LC, IgA, IgG or IgM heavy chain staining (Fig. 2). Examination of a skin specimen under electron microscopy revealed slightly electron-dense amorphous non-fibrillar deposits in the upper part of the papillary dermis, just below the basement membrane zone, and no amyloid fibrillar deposits (Fig. 3).
Fig. 2. Direct immunofluorescence. (a) Abundant granular lambda light chain deposits at the dermoepidermal junction and in the papillary dermis, with (b) no kappa light chain deposits (×400 magnification).
Fig. 3. Presence of slightly electron-dense granular non-fibrillar deposits in the upper papillary dermis. *Basal keratinocytes. Dashed line: basement membrane zone; arrows: abnormal deposits) (transmission electron microscopy, ×30,000 magnification).
A diagnosis of LCDD with cutaneous and renal involvement complicating a smouldering multiple myeloma was made and a chemotherapy was started in March 2015. First-line treatment, with a combination of bortezomib, cyclophosphamide and dexamethasone, had no effect on serum FLC or paraprotein level, cutaneous disorders and nephrotic syndrome. Second-line treatment with lenalidomide, cyclophosphamide and dexamethasone was also ineffective. Third-line treatment with a combination of bortezomib, bendamustine and dexamethasone was ongoing when the patient died suddenly at home in February 2016.
A variety of skin disorders are associated with MG. In some cases of MGCS, such as scleromyxoedema (3) or neutrophilic dermatosis (4), the pathogenesis is uncertain. Presumptions exist regarding the role of the monoclonal component in the pathogenesis of the skin disease, because these skin manifestations are highly associated with the presence of a paraprotein, but the link is not yet understood.
In other skin disorders, such as amyloidosis (5) or linear IgA bullous dermatosis (6), the pathogenesis is well identified and the skin changes are the result of monoclonal immunoglobulin deposition or its antibody activity.
In bullous amyloidosis, the blistering is believed to result from fragility of amyloid-laden dermal connective tissue (2). Alternatively, in few cases of non-amyloidosis bullous dermatosis associated with MG, the monoclonal immunoglobulin is an auto-antibody directed against skin components (7).
In MGCS, the demonstration of the direct role of the monoclonal component in skin injury is crucial, as it has therapeutic implications and should lead to commencement of chemotherapy targeting the plasma clone.
It is important to note that LC deposits are not detected with routine DIF using anti heavy chain antibodies and can be missed if not specifically looked for, using specific staining for kappa and lambda LC. Electron microscopic (EM) examination can also be useful to assess LC granular deposits in the skin. Detection of a monoclonal component in serum or urine is a strong argument to perform such investigation. Immuno-EM would have been interesting, but could not be performed on site. However, the combination of light microscopy immunofluorescence coupled with electron microscopy studies, showing non-organized linear LC deposits along the basement membrane is usually sufficient to make a diagnosis of LCDD.
In conclusion, we describe here the first case of bullous LCDD mimicking BP, extending the spectrum of entities forming MGCS. It is important to determine any form of skin involvement in LCDD. Beyond the fact that it will enable commencement of accurate treatment of the skin injury, it can help in making an early diagnosis before systemic deposition and evolution to end-stage kidney disease.


 Click to show fullsize
Click to show fullsize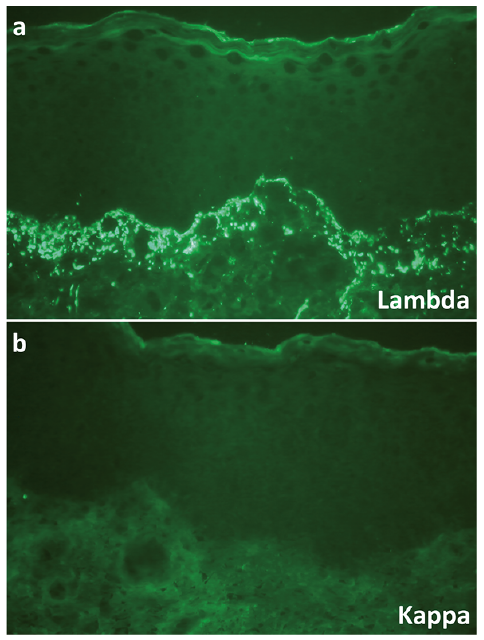 Click to show fullsize
Click to show fullsize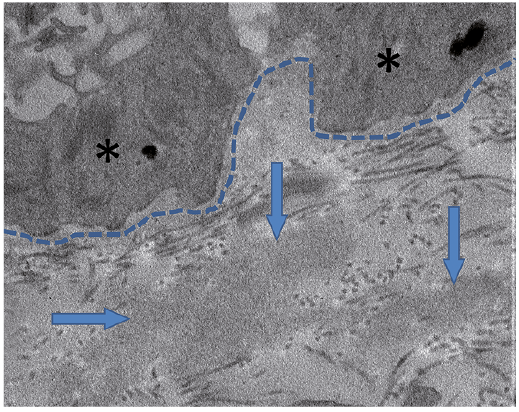 Click to show fullsize
Click to show fullsize