


Departments of 1Dermatology and Allergy Centre, 2Clinical Pathology and 4Clinical Genetics, Odense University Hospital, DK-5000 Odense, Denmark, and 3Institute for Human Genetics, Medical Center – University of Freiburg, Freiburg, Germany. E-mail: Kristine_pallesen@hotmail.com
Accepted Aug 23, 2017; Epub ahead of print Aug 23, 2017
Ichthyosis with confetti (IWC), also known as congenital reticular ichthyosiform erythroderma (CRIE) or ichthyosis variegata, is an autosomal dominant form of congenital ichthyosis. It is extremely rare, with fewer than 50 cases reported in the world literature (1–7).
Children are born as collodion babies or with ich-thyosiform erythroderma. Later in childhood, the classic confetti-like spots of healthy-looking wild-type skin develop due to revertant mosaicism, which are clues to the diagnosis. Histopathological examination of a skin biopsy will show very characteristic vacuolar changes of the keratinocytes in the superficial epidermis. The diagnosis can be confirmed by genetic analysis.
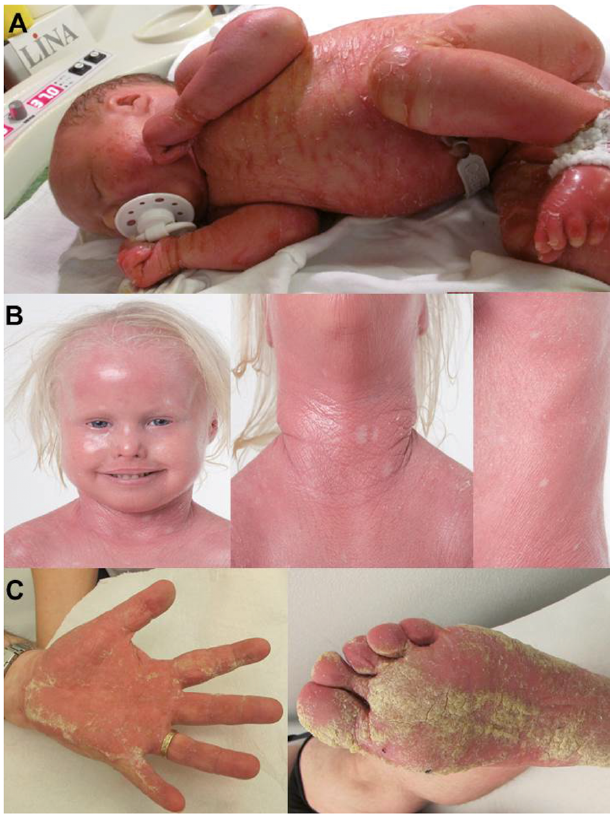
A collodion baby (Fig. 1A) was born at term. She persistently had scaly erythroderma, palmoplantar keratoderma, nail dystrophy and malformed pinnae, ectropion and mammillae hypoplasia. At the age of 2 years it was evident that she had hypotrichosis on the scalp, sparse eyebrows and eyelashes, and moderate hypertrichosis on the body. At the age of 3 years small pale confetti-like spots appeared on her skin (Fig. 1B). She was persistently growth retarded, with length approximately –2 standard deviations (SD) and weight approximately –3 SD. Sequencing of genomic DNA extracted from peripheral blood lymphocytes demonstrated a KRT10 mutation: c.1374-1G>A in intron 6, in heterozygous form.
Fig. 1. Clinical features. (A) Index patient who presented as a collodion baby at birth and (B) 3 years later with ichthyosis with confetti. (C) Mosaic father presenting with Blaschko-linear verrucous and patchy hyperkeratosis as well as palmoplantar keratoderma. A written permission from the parents is given to publish these photos.
The patient’s father was 45 years of age, and had acral linear hyperkeratosis and palmoplantar keratoderma (Fig. 1C). In childhood he had been wrongly diagnosed and published as having incontinentia pigmenti (8); however, at that time no genetic data were available. Later on the following diagnoses were considered based on clinical and histopathological features: naevus unius lateris, ichthyosis hystrix, and linear porokeratosis. His skin at birth was described by his parents to be oedematous with occasional blistering. He subsequently developed palmoplantar keratoderma and patchy keratotic plaques and Blaschko-linear verrucous hyperkeratosis on his extremities.
Several skin biopsies from the father had been taken over the years from the verrucous, linear lesions and the palmoplantar keratoderma. Some of these biopsies were available for review, and showed essentially similar changes, such as features of porokeratosis (papillomatosis, hyperkeratosis with cornoid lamellar changes and vacuolar changes of keratinocytes in the decreased granular layer), as well as features of epidermolytic hyperkeratosis/ichthyosis hystrix (vacuolar changes of keratinocytes in the upper spinous zone, but not confluent and without clumping of keratohyalin granules). The histo-pathology, however, was not diagnostic for either of these conditions.
A skin biopsy from the daughter revealed the same characteristic vacuolar changes of the superficial keratinocytes, but with much less hyperkeratosis. No definitive diagnosis was made.
A diagnosis of IWC was suggested, based on clinical background, and the biopsies from both patients were reviewed. The changes in the biopsies were found to be identical and diagnostic of IWC (Fig. S1).
Mutation analysis was performed on DNA extracted from additional biopsies from affected and unaffected skin of the father, which showed the same KRT10 mutation c.1374-1G>A in a mosaic state. The mutation was not present in DNA extracted from peripheral blood lymphocytes from the father.
IWC belongs to the subgroup of keratinopathic ich-thyoses within the group of non-syndromic ichthyoses. According to the most recent nomenclature, the disease name should be ichthyosis variegata (2), which is, how-ever, not as descriptive as IWC. The newborn either presents as a collodion baby with a parchment-like membrane covering its body surface or with ichthyosiform erythroderma. Other manifestations are hypoplastic mammillae, ear deformities, hypertrichosis, palmoplantar keratoderma and short stature. The clinical hallmarks are small confetti-like spots of normal skin that develop during early childhood and puberty. The number and size of spots increase over time.
The confetti-like spots of normal skin are previously demonstrated to be caused by reversion of mutations of KRT10 or KRT1 by mitotic recombination (9, 10).
The precise diagnosis of the index patient became apparent when she developed islands of normal skin (revertant mosaicism), and was confirmed by genetic testing. Furthermore, the characteristic vacuolar changes of the superficial keratinocytes were identified histologically, when former biopsies were revised (11). The diagnosis was confirmed at the molecular-genetic level, as a previously reported KRT10 acceptor splice site mutation in intron 6 was demonstrated (9, 12).
The present family is unique, as they present IWC in 2 generations with clinically different phenotypic presentations from the same mutation. The father presented a phenotype resembling naevoid epidermolytic ichthyosis (epidermolytic hyperkeratosis) and the girl presented a clear phenotype of IWC (1, 2, 12). Epidermolytic ich-thyosis is also caused by mutations in KRT10, although not the keratin tail, which is affected in IWC. Histo-pathologically, however, the characteristic vacuolar changes in the superficial keratinocytes were similar in the father and the daughter, diagnostic of IWC and ruling out other variants of keratinopathic ichthyoses.
Since both the daughter and the father carried the same KRT10 splice site mutation: c.1374-1G>A in intron 6, it was obvious that the father carried the mutation in at least some of his germ cells. The father was a mosaic based on a postzygotic mutation. The identification of a germline mosaicism in the father has implications for the risk of an affected child in future pregnancies, and for the genetic counselling of the family. Prenatal diagnosis can be offered in future pregnancies.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize