


Department of Dermatology, University of Tokyo Graduate School of Medicine, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8655, Japan. *E-mail: miyagawat-der@h.u-tokyo.ac.jp
Accepted Sep 19, 2017; Epub ahead of print Sep 20, 2017
Atrophic dermatofibrosarcoma protuberans (DFSP) is a very rare variant of DFSP, which presents as an atrophic, asymptomatic plaque that can be difficult to diagnose. Like classic DFSP, standard wide excision is necessary for the treatment of atrophic DFSP, thus accurate diagnosis is indispensable. We report here a case of atrophic DFSP that presented as an unprecedentedly large atrophic plaque on the anterior chest, which was diagnosed and treated successfully.
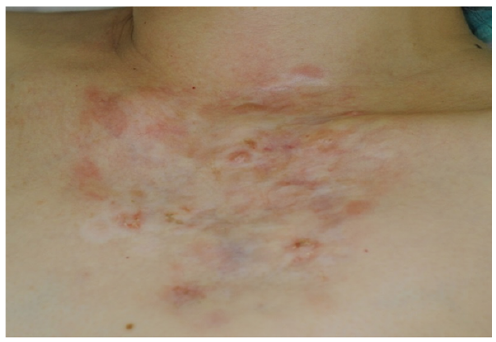
A 48-year-old healthy Japanese woman presented with an 8-year history of red nodules surrounded by a palm-sized (13×12 cm) area of red atrophic plaque on the anterior chest wall (Fig. 1). She had no history of surgery or trauma to the chest. She had noticed the atrophic plaque at the age of 40 years. The plaque expanded gradually, followed by development of red nodules. The results of laboratory investigations, including anti-nuclear antibody, were within normal limits. Histological analysis showed an extensive basophilic nodular area in the dermis and upper hypodermis in addition to dermal and epidermal atrophy (Fig. 2A). This nodular area was composed of monomorphic spindle cells with minimal cellular atypia, which were arranged in a characteristic storiform pattern (Fig. 2B). Immunohistochemical staining revealed that these spindle cells stained positively for CD34 (Fig. 2C) and negatively for S100 protein or epithelial membrane antigen (EMA), which suggested a diagnosis of DFSP, plaque-like CD34-positive dermal fibroma or solitary fibrous tumour. To confirm the diagnosis, we further performed reverse transcriptase PCR (RT-PCR) according to the method reported previously (1). We detected collagen type 1-α-1 (COL1A1)-platelet-derived growth factor β-chain (PDGFB) fusion gene (Fig. 2D), and a final diagnosis of DFSP was made. The patient was treated with a wide excision followed by split-thickness skin grafts. Local excision was performed with lateral safety margin of 3 cm. No recurrence was found at 10-month follow-up.
Fig. 1. A palm-sized red atrophic plaque with red nodules on the anterior chest.
Fig. 2. Histopathology, immunostaining and molecular analysis. (A, B) Haematoxylin-eosin staining of the atrophic plaque lesion (A, ×12.5; B, ×400). (C) CD34 immunostaining of the atrophic lesion (×400). (D) The detection of COL1A1–PDGFB gene fusion transcripts in the skin samples from the lesion.
Atrophic DFSP is a very rare variant of DFSP. DFSP typically develops as an initial plaque in the early stage followed by a nodule in the late stage (2). The initial plaque could be atrophic and persistent in rare cases, called atrophic DFSP (3). Atrophic DFSP was first reported in 1985 (3), and to date there have been 45 reported cases of this variant, including our case. Demographically, truncal involvement is more common in atrophic DFSP (75% of cases including the shoulder region) than in typical DFSP (50–60% of cases), and the sex distribution of atrophic DFSP shows a slight female predominance in the atrophic variant (63%), while that of typical DFSP is relatively equivalent (4). Although the mean age of onset for atrophic DFSP is 29 years (4), it can arise in childhood or congenitally (5–8).
Clinically, atrophic DFSP presents as an atrophic, asymptomatic plaque that can be difficult to distinguish from morphoea, idiopathic atrophoderma, atrophic scar, anetoderma or lipoatrophy (4). Due to its non-protuberant appearance, it is difficult to make a diagnosis of DFSP at first sight. Tumour size has been reported to range from 0.5 to 8 cm in greatest diameter (4); however, our case had a diameter of 13 cm. In relation to disease duration, a case was reported in which a non-protuberant, morphoea-like lesion remained for 20 years before prominent nodules developed (9). Therefore, biopsy should be considered when atrophic lesions occur in the skin without an obvious cause, regardless of size and duration.
Histologically, atrophic DFSP is characterized by dermal atrophy in addition to monomorphic spindle cells with minimal atypia arranged in a storiform pattern (2, 10). The dermal thickness is usually reduced to less than 50% in the middle of the tumour (10), and epidermal atrophy can be observed in some cases (11). The infiltrate of spindle cells usually extends to the hypodermis. Histological differential diagnosis includes atrophic dermatofibroma, atypical fibroxanthoma and undifferentiated pleomorphic sarcoma (12). Although these tumours are composed of spindle-shaped cells, atrophic dermatofibroma and atypical fibroxanthoma usually affect only the dermis, and undifferentiated pleomorphic sarcoma consists of pleomorphic spindle cells with frequent mitoses having some necrotic areas in the lesion (2). Immunohistochemistry is also helpful in distinguishing atrophic DFSP from these tumours. Atrophic DFSP is strongly positive for CD34, while these tumours are negative (10, 13, 14). Atrophic DFSP shows no reactivity to S100 protein, EMA, desmin or smooth muscle actin, which is another aid in diagnosing this tumour.
Atrophic DFSP behaves like classic DFSP in both genetics and prognosis (2, 8), and therefore standard wide excision is necessary for the treatment of atrophic DFSP. It is advised that standard excision should be performed with a lateral safety margin of 3 cm (15). Imatinib therapy can be considered when the presence of COL1A1–PDGFB fusion gene is suggested by RT-PCR or fluorescence in situ hybridization (15).
In summary, an atrophic lesion with no apparent cause can be a sign of DFSP, and despite its rarity it is important to consider biopsy in cases with such lesions.


 Click to show fullsize
Click to show fullsize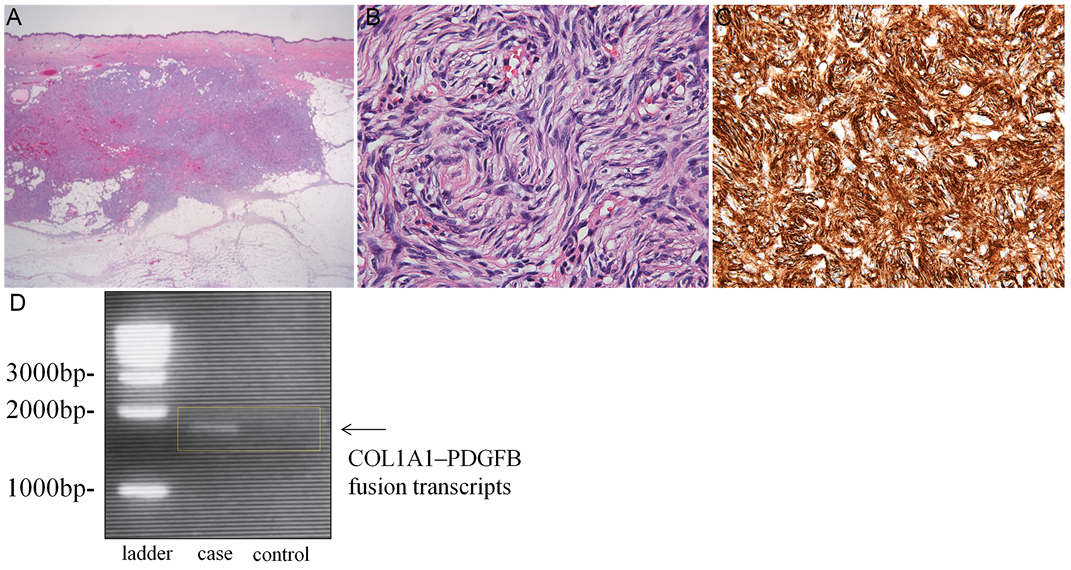 Click to show fullsize
Click to show fullsize