


1Department of Dermatology, Venereology and Allergology, and 2Department of Pathomorphology, Wroc?aw Medical University, Cha?ubi?skiego 1, PL-50-368 Wroc?aw, Poland. E-mail: magda.zychowska@gmail.com
Accepted Sep 27, 2017; Epub ahead of print Sep 27, 2017
Primary cutaneous CD30+ lymphoproliferative disorders comprise 3 entities: lymphomatoid papulosis (LyP), primary cutaneous anaplastic large cell lymphoma (pc-ALCL) and borderline diseases (1). LyP is characterized by the presence of recurrent and self-healing papulo-necrotic lesions. This entity may be associated with another lymphoproliferative disease, e.g. pc-ALCL, mycosis fungoides or Hodgkin’s lymphoma, in 20% of cases (1, 2). Pc-ALCL predominantly affects older men and presents as a solitary rapidly growing nodule or tumour in 80% of patients. The disease is characterized by excellent prognosis with 10-year disease-related survival rates of 90% (1, 2).
LyP and pc-ALCL are currently considered to be opposite ends of a spectrum of the same disease. Differentiation of these conditions should be based on clinical presentation and on the course of the disease, as histopathology may not be decisive (1). In particular, LyP type C, which is characterized by nodular infiltration of large atypical lymphoid cells, bears a strong microscopic resemblance to pc-ALCL (1, 2).
The immunophenotypical hallmark of LyP and pc-ALCL is the expression of CD30, a transmembrane protein of the tumour necrosis factor (TNF)-receptor superfamily, which was found to be associated with the production of cytokines participating in humoral responses (3). Caproni et al. (4) found that remarkable numbers of CD4+ T lymphocytes in lesional skin of patients with active atopic dermatitis (AD) expressed CD30. It is suspected, that clonal transformation of CD30+ T cells in inflammatory infiltrates may lead to the development of primary cutaneous CD30+ lymphoproliferative disorder.
We present here a patient with long-lasting severe AD, who developed LyP type C, and several months later pc-ALCL, with subsequent nodal involvement.
A 59-year-old woman presented to the Department of Dermatology in 2015 with fast-growing nodules on both lower legs. She had had severe, persistent AD since infancy. Over the years, she had been treated with several systemic therapies, including 6 cycles of oral photochemotherapy, performed 3 times per week (ultraviolet A (UVA) doses started at 1.5 J/cm2; cumulative UVA dose 320 J/cm2) and several courses of oral prednisone in periods of erythrodermic exacerbation of AD. Oral cyclosporine A (CsA) had been introduced 3 times. In 2004, CsA was stopped after 2 weeks due to nausea and vomiting. In 2008, the drug was discontinued after several weeks because of hypertension. CsA, 2.5 mg/kg/day, was introduced again in 2010; however, with no satisfactory response after 10 weeks of treatment. In addition, the patient had used numerous antihistamines over the years and topical treatment was based on long-term use of corticosteroids and calcineurin inhibitors.
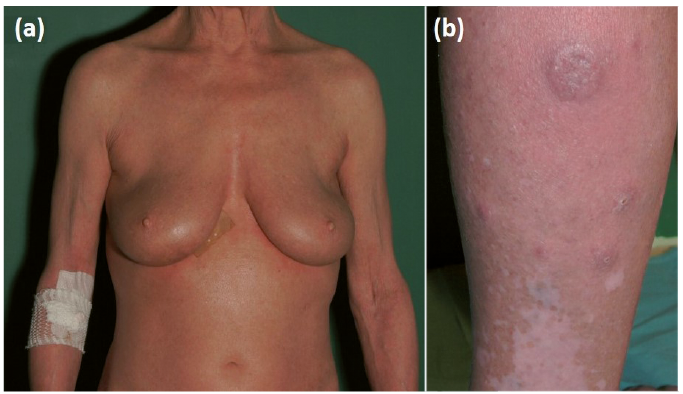
Five months prior to hospitalization, red papules and nodules up to 1 cm in diameter developed on the anterior surface of the patient’s left thigh. Histopathological examination of one of the lesions revealed dense lymphocytic infiltration, which was positive for CD30, CD15, MUM-1, CD2 and CD45RO, but not for CD20, CD3, CD4, CD8, CD56, ALK-1, or EBV. The proliferation index Ki67 was expressed in more than 90% of cells. The lesions disappeared after several days of application of potent topical corticosteroid. Following clinicopathological discussion, a diagnosis of LyP type C was established. One month prior to the patient’s hospitalization, tense nodules started to develop on both lower legs and rapidly increased in diameter. On admission, clinical examination revealed extensive lichenified eczema (Fig. 1a) and several tense pinkish nodules and ulcerated tumours on the lower legs (Fig. 1b). Blood cell count and renal and hepatic parameters were within the normal ranges. Extended laboratory investigations revealed elevated level of lactate dehydrogenase (279 U/l; normal range 125–220 U/l), β2-microglobulin (3 mg/l; normal range 1.09–2.53 mg/l) and total IgE (37,500 IU/ml; normal range 100 IU/ml). A biopsy from an eczematous skin revealed acanthosis, focal hyperkeratosis and scarce perivascular lymphocytic infiltrate in the upper dermis (Fig. S1a), which was immunophenotypically CD3+, CD4+, CD8+, CD20–. Only single medium-sized to large cells were positive for CD30. Ki67 was expressed in 15% of cells. Histopathological examination of one of the nodules on the lower legs showed dense lymphocytic infiltration of large anaplastic cells, which were positive for CD4, CD30, MUM-1 and negative for CD8, CD20, ALK-1. Ki67 was expressed in 90% of cells (Fig. S1b–h). No abnormalities were detected on computed tomographic scans of the chest, abdomen and pelvis. A diagnosis of pc-ALCL was made. According to the European Organisation for Research and Treatment of Cancer (EORTC), International Society for Cutaneous Lymphomas (ISCL) and United States Cutaneous Lymphoma Consortium (USCLC) consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders (5), therapy with methotrexate 20 mg/week in combination with psoralen plus ultraviolet A (PUVA) was initiated. A gradual improvement in eczematous lesions and a reduction in the diameter of the tumours was achieved. Seven months after initial diagnosis, rapid enlargement of the inguinal lymph nodes was observed on clinical examination. Histopathology of the right inguinal lymph node confirmed lymph node involvement with anaplastic large cell lymphoma, with the immunophenotype positive for CD4, CD30, and MUM-1, negative for CD3, CD8, CD20, and ALK-1, and Ki67 positive in 70–80% of neoplastic cells. The patient received CHOP (cyclophosphamide, doxorubicin, vincristine sulphate, prednisone) chemotherapy with complete remission of the disease. She has remained symptom-free for one year.
Fig. 1. Clinical presentation. (a) Extensive lichenified eczema with a tendency to erythroderma; (b) Tense nodules and ulcerated tumours on the lower legs.
According to Vajdic et al. (6), patients with AD have a 2-fold increased risk of developing T-cell lymphoma, particularly mycosis fungoides and Sézary syndrome. The relationship between AD and primary cutaneous CD30+ lymphoproliferative disorders appears to be even more complex. So far, 9 cases, including one case series of 4 patients, of LyP (n = 3) or pc-ALCL (n = 6) in adult patients with persistent AD have been reported in the English literature (7–12). Four of the 9 patients have been treated with CsA during or prior to the development of lymphoproliferative disease. There was a male preponderance (M:F 7:2) and mean age at onset was 35.6 years. Although pc-ALCL is considered a disease of relatively good prognosis with 10-year survival rate of 90%, in the reported AD patients the course was progressive, resulting in death in 2 cases (8, 11) (Table SI).
Patients with AD have been found to have an increased number of CD30+ lymphocytes in lesional skin, which may explain why LyP and pc-ALCL developed in relatively young patients with severe AD since early childhood (4). Nevertheless, it does not account for more aggressive course with a tendency to systemic involvement in these patients.
The pathogenesis of CD30+ lymphoproliferative disorders remains unclear. Pc-ALCL is not rare in organ-transplant recipients and HIV-infected individuals (2). In patients with AD, clonal transformation of CD30+ T cells is probably a complex and multifactorial process. Several aspects need to be taken into consideration as potential inducing factors, including immunosuppressive treatment with CsA, photochemotherapy-provoked malignancy and chronic stimulation of lymphocytes by superantigens (9). The lymphoproliferative risk associated with CsA treatment was particularly underscored by Kirby et al. (7), who were the first to report a case of CD30+ lymphoproliferative disease in a patient with severe AD. In the aforementioned patient, LyP developed during monotherapy with low-dose CsA and completely remitted after drug withdrawal. Nevertheless, it should be taken into consideration, that 2 patients with AD, reported so far, who developed very aggressive pc-ALCL with fatal course, had never been treated with CsA or other immunosuppressive agent (8, 11). Another disputable issue is the safety of long-term use of systemic antihistamines and topical calcineurin inhibitors. So far, the potential link between primary cutaneous marginal zone B-cell lymphoma and chronic intake of antihistamines has been underscored by several authors (13, 14). The association between malignancy and topical calcineurin inhibitors remains questionable and has many supporters, despite a growing body of evidence confirming the safety of long-term use of tacrolimus and pimecrolimus in patients with AD (15).
In the presented patient, the initial papulo-nodular lesions on the thigh were followed several months later by the development of rapidly growing tumours on both lower legs. Despite similar histological and immuno-phenotypical findings, we diagnosed the initial lesions to be LyP type C, and the fast-growing tumours pc-ALCL. The development of LyP and second lymphoproliferative disease in one patient is not rare. Moreover, LyP type C and pc-ALCL may be histologically indistinguishable and the clinical presentation is crucial for making the final diagnosis. Considering the size of the initial lesions (papules and nodules up to 1 cm diameter) and rapid disappearance after application of topical corticosteroids, we are in favour of the diagnosis of LyP type C. Likewise, the rapidly growing large tumours on lower legs were diagnosed to be pc-ALCL.
We believe that the co-occurrence of severe AD and CD30+ lymphoproliferative disorders in the presented patient was not coincidental. Patients with long-lasting AD should be examined regularly for newly developed nodular lesions, which may raise suspicion of lym-phoproliferative disease.


 Click to show fullsize
Click to show fullsize