


1Division of Dermatology, 2Division of Hematology, Department of Internal Medicine, Kobe University Graduate School of Medicine, 6500017 5-2, Kusunoki-cho7, Chuo-ku, Kobe-shi, Hyougo, Kobe, 3Department of Dermatology, Hirosaki University Graduate School of Medicine, Hirosaki, 4Department of Microbiology, Kansai Medical University, Hirakata, and 5Department of Haematology, Nishiwaki Municipal Hospital, Nishiwaki, Japan. E-mail: aiichi39@gmail.com
Accepted Oct 19, 2017; Epub ahead of print Oct 23, 2017
Erythropoietic protoporphyria (EPP) is an inherited disorder caused by partial deficiency of ferrochelatase (FECH), the last enzyme in the heme biosynthetic pathway. The main clinical symptoms are oedematous erythema or blisters with itching and pain after exposure to sunlight, and liver dysfunction due to erythrocytolysis and accumulation of porphyrin. Erythropoietic protoporphyria is generally diagnosed during childhood; however, some adult-onset cases secondary to myelodysplastic syndrome (MDS) have been reported. Acquired EPP can develop in an individual with the occurrence of a chromosome abnormality in the FECH due to myelodysplastic syndrome, which results in reduced expression of ferrochelatase. In the case described here, the methyltransferase inhibitor, azacitidine (AZA), suppressed progression of MDS as well as the symptoms of photosensitivity, indicating that it may be effective in reversing FECH gene silencing.
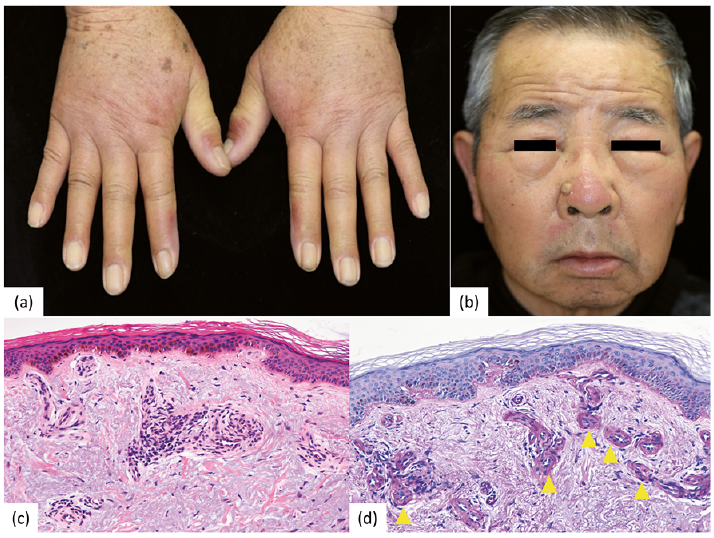
A 78-year-old man visited our hospital with pain and swelling of his hands and face due to sun exposure since 4 months earlier. He had been treated with an oral angiotensin II receptor blocker, telmisartan, for high blood pressure until 1 year before the hospital visit. He initially exhibited oedematous changes on the dorsa of both hands, with lichenification extending from the dorsa to the forearms and greyish-red oedematous changes on the dorsum of the right hand (Fig. 1a). Diffuse faint erythema was observed on the dorsum of the nose and the interior of the nasolabial fold (Fig. 1b). Using a slide projector equipped with a 250-W halogen lamp illuminating at wavelengths of between 380 and 780 nm and situated 15 cm away from the patient’s back, a poorly demarcated, non-pruriginous, erythematous macule appeared after 15 min irradiation. The patient’s skin phototype was type IV, based on the classification by Thomas B. Fitzpatrick,
Fig. 1. Clinical images and histopathological findings. (a) Obvious lichenification is seen from the dorsa of both hands to the forearms as well as oedematous and erythematous changes. (b) Skin on the patient’s face shows diffuse erythema on the dorsum of the nose/nasolabial fold. (c) Haematoxylin-eosin stain (magnification ×160): Hyperpigmentation is seen in the basal epidermal layer. Amorphous eosinophilic material is deposited around the vessels in the papillary and lower layers of the dermis. (d) Periodic acid-Schiff (PAS) stain (magnification ×320): PAS-positive tissue with deposits of amorphous material. Permission is given by the patient to publish these photos.
Blood examination showed moderate macrocytic anaemia (red blood cell (RBC) count 2.6 million/µl (normal 4.3–5.7); haemoglobin (Hb) level 9.1 g/dl (13.5–17.5); haematocrit 27.6% (34.8–25.0); mean corpuscular volume 106 fL (80–100); mean corpuscular Hb level 35 pg (30–35); and mean corpuscular Hb concentration 33% (30–35)). Biochemical examination showed increased protoporphyrin RBC levels (1,341 µg/dl, normal 30–86) and normal liver function. Urinalysis was normal. Histopathological examination with haematoxylin–eosin staining of skin biopsies from the dorsa of the right hand showed basal pigmentation, with periodic acid-Schiff-positive eosinophilic amorphous depositions around vessels in the upper dermis (Fig. 1c, d). Acquired EPP was suspected on the basis of the late onset of symptoms and moderate macrocytic anaemia.
Bone marrow examination showed normoplastic marrow and erythroid hyperplasia with dysplastic features.
G-banding and spectral karyotyping of the marrow cells showed an abnormality in the chromosome 18q21 in which the FECH gene resides (Fig. S1a). The patient’s symptoms were consistent with refractory anaemia with ring sideroblasts because bone marrow examination showed erythroid dysplasia, the ratio of ringed sideroblasts was 26%, and blood examination showed anaemia at his first visit only.
The FECH activity in the peripheral lymphocytes of the patient was 3.9 ± 0.022 nmol/mg protein/h, which was higher than that of healthy controls (n = 2) (2.8 ± 0.6 nmol/mg protein/h).
Multiplex ligation-dependent probe amplification (MLPA) indicated that fibroblast cells derived from the patient’s skin had the same number of FECH genetic copies as controls; however, these numbers in the patient’s bone marrow cells were decreased to 60–70% of controls (Fig. S1b).
Regarding genetic typing, the genotype of SNP was IVS3-48C/T in fibroblast cells, and alleles with IVS3-48C were the main constituents in bone marrow cells (Fig. S1c).
Two months of subcutaneous administration of an erythropoiesis-stimulating agent (ESA), darbepoetin alfa (240 µg/week) resulted in significant improvements in anaemia, but an increase in porphyrin levels (Fig. S2). We assumed this to be the result of porphyrin accumulation via an increase in heme metabolism. For hepatic support, oral glycyrrhizin and ursodeoxycholic acid had been administrated for approximately 1 month, during which time liver enzyme levels normalized rapidly.
Because MDS progressed to RAEB-2 (refractory anaemia with excess blasts) and became RBC-transfusion dependent, intravenous AZA administration was initiated 10 months after the initial visit, at a dose of 50 mg/m2 daily for 5 days every 28 days, in addition to ESA. After 9 courses of AZA administration, the ratio of blasts in the bone marrow was well controlled and the chromosome karyotype appeared 46XY, normal in G-banding. Furthermore, protoporphyrin levels improved and his photosensitivity did not recur after the initiation of AZA treatment.
In our case, a chromosomal anomaly was found in the FECH gene on chromosome 18q21.31. FECH copy numbers were decreased and the FECH gene mainly showed the IVS3-48C in bone marrow cells and IVS3-48C/T in fibroblasts. We assumed that the original genotype of IVS3-48 in the patient was C/T and that the chromosome abnormality in the 18q21.31 region on the IVS-48T allele occurred secondarily, resulting in lower expression of the FECH protein and leading to EPP development. The FECH activity in the peripheral lymphocytes was of a similar level to that of healthy controls, probably because FECH in the erythroblasts, and not lymphocytes, had harboured an aberrant clone.
We found 19 reported cases of late-onset EPP associated with MDS (1–132). The mean age was 60.7 years, and 94.1% of the cases were diagnosed with EPP following the onset of photosensitivity. The mean blood protoporphyrin level of all of these patients was 4391.4 µg/dl RBCs (normal < 80).
Erythropoiesis-stimulating agents (ESA) are administered to low-risk patients with MDS who are ineligible for transplantation. EPP symptoms, such as hepatic disorder development or photosensitivity, worsen owing to porphyrin accumulation due to the facilitation of heme metabolism. The methyl transferase inhibitor AZA that targets epigenetic changes in MDS is appropriate in high-risk patients with MDS for haematological remission and improvement. Nishikawa et al. (13) reported a patient with MDS and EPP treated with AZA whose hepatic disorder and photosensitivity reaction did not worsen for one year following the initial administration of AZA.
Although we initiated ESA treatment in our case, we reduced the dose and extended the administration interval owing to the increase in protoporphyrin levels. The progression of MDS necessitated a blood transfusion and AZA administration. We assumed that early full-dose ESA administration caused liver dysfunction due to increased protoporphyrin levels in individual erythroblasts. After initiation of AZA treatment, protoporphyrin levels gradually decreased, with control of Hb levels, and stabilization of liver function.
Patient reactions to ESAs vary according to the quality and quantity of blood cell systems in MDS states. On initiating AZA administration, we observed no further MDS progression up to 9 courses in total, along with the ESA and blood transfusions, and porphyrin levels improved. These results show that administration of ESA may increase porphyrin levels, which is beneficial for improving anaemia with careful monitoring of liver function and porphyrin levels. In addition, AZA can indirectly prevent EPP worsening by suppressing the progression of MDS. 5-azacitidine may also be effective in reversing FECH gene silencing caused by DNA methylation.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize