


1University François Rabelais Tours, 2CHRU Tours, Department of Dermatology, 3CHRU Tours, Clinical Investigation Center-INSERM 1415, Tours, 4Department of Dermatology, University Hospital Center of Toulouse, Toulouse, 5Department of Dermatology and Reference Center for Genodermatoses and Rare Skin Diseases (MAGEC), University Hospital Necker-Enfants Malades, Paris, Departments of Dermatology, 6University Hospital Center of Angers, Angers, 7University Hospital Center of Nice, Nice, 8University Hospital Center of Reims, Reims, 9Hospital Center of le Mans, le Mans, 10University Hospital Center of Dijon, Dijon, 11University Hospital Center of Rouen, Rouen, 12Hospital Center of Quimper, Quimper, 13University Hospital Center of Montpellier, Montpellier, 14University Hospital Center of Nantes, Nantes, 15University Hospital Center of Amiens, Amiens, 16University Hospital Center of Rennes, Rennes, and 17INSERM U930, CHRU Tours, Laboratory of Biochemistry and Molecular Biology, Tours, France
#These authors contributed equally and should be considered as first authors.
Patients with an inherited autosomal-dominant disorder, capillary malformation–arteriovenous malformation (CM-AVM), frequently have mutations in Ras P21 protein activator 1 (RASA1). The aims of this study were to determine the prevalence of germline RASA1 variants in a French multicentre national cohort of children, age range 2–12 years, with sporadic occurrence of capillary malformation (CM) of the legs, whatever the associated abnormalities, and to identify genotype–phenotype correlates. DNA was extracted from leukocytes in blood samples, purified and amplified, and all exons of the RASA1 gene were analysed. Among 113 children analysed, 7 had heterozygous variants (6.1%). Four different variants were identified; 2 were new. In children with RASA1 variants, CMs were more frequently bilateral and multifocal. In conclusion, RASA1 variants are rarely found in children with sporadic CM of lower limbs without CM-AVM syndrome. CMs in this study were heterogeneous, and no disease-causing relationship could be proven.
Key words: RASA1; mutations; variants; polymorphism; capillary malformations; vascular anomalies.
Accepted Nov 2, 2017; Epub ahead of print Nov 7, 2017
Acta Derm Venereol 2018; 98: XX–XX.
Corr: Annabel Maruani, CHRU Tours, Hospital Trousseau, Department of Dermatology, Avenue de la République, FR-37044, Tours Cedex 9, France. E-mail: annabel.maruani@univ-tours.fr
Our knowledge of the genetic causes of vascular malformations has increased considerably in recent years. In capillary malformations (CMs), post-zygotic activating mutations of G protein subunits alpha Q (GNAQ) and alpha 11 (GNA11) have been found in affected skin of patients with facial port-wine stains in Sturge-Weber syndrome or phacomatosis pigmentovascularis (1, 2). Postzygotic activating phosphatidylinositol-4,5-bisphosphate 3 catalytic (PIK3CA) mutations also account for a number of capillary/lymphatic malformations as part of the CLOVES and macrocephaly-CM syndromes (3, 4). Cutaneous CMs might be associated with germline mutations in the Ras P21 protein activator 1 gene (RASA1) as part of the autosomal dominant syndrome, capillary malformation-arteriovenous malformation (CM-AVM) (5, 6).
CM-AVM syndrome was initially described as a combination of very suggestive and typical multifocal cutaneous CMs (small, round CMs, with pale halos), associated with high-flow superficial lesions, arteriovenous fistulas (AVFs) or cerebral AVMs (6). However, mutations in RASA1 have been reported in 3 families of patients with CM, but without AVM (7). Similarly, Wooderchak-Donahue reported RASA1 mutations associated with hereditary CM, with or without AVF or AVM in 8 subjects (8). In sporadic cases, germline RASA1 mutations have been identified in the peripheral blood of 1 patient with Sturge-Weber syndrome (9).
RASA1 mutations were found to be responsible for aberrant lymphatic architecture and functional abnormalities other than CM in mice and humans (10). RASA1 encodes the 120-RasGAP protein involved in the RAS-MAPK signalling pathway that controls proliferation, migration, and survival of various cell types, including vascular endothelial cells. The P120-RasGAP protein converts active GTP-bound Ras into inactive GDP-bound Ras and has been involved in vascular development in RASA1-/- mice (11). Loss of function mutations in RASA1 in humans are associated with abnormal angiogenic remodelling of the primary capillary plexus, with vascular anomalies, not compensated by other RasGAPs, such as RASA2, RASAL, or NF1 (6, 12).
Since 2010 we have implemented a multicentre national cohort (CONAPE) from 14 French tertiary centres of paediatric dermatology specialized in vascular anomalies, including 120 children aged 2–12 years with CMs of the legs. CMs are either simple, combined (associated with a venous, arterial and/or lymphatic abnormal component), or associated with other anomalies, as defined in the updated classification of vascular anomalies (13).
The aim of this study was to assess the prevalence of RASA1 variants in children with CM of the legs from this cohort, and to identify genotype-phenotype correlates.
Data were obtained from a multicentre French national cohort including 120 children with CM of the legs in 14 French hospitals (Departments of Dermatology from Amiens, Angers, Dijon, Le Mans, Montpellier, Nantes, Nice, Paris-Necker, Quimper, Reims, Rennes, Rouen, Toulouse, Tours). The recruitment period ranged from 10 November 2010 to 8 January 2015.
Eligibility criteria were age 2–12 years with congenital CM of at least 1 lower limb, regardless of the size and type of CM and associated anomalies.
Both parents gave their informed consent for children to participate in the study. The protocol was approved by the institutional review board of the University Hospital Center of Tours (#2010-R22).
At baseline, dermatologists collected data on demographics (age, sex), anatomical measurements, personal and familial medical history, clinical description of the CM (location, features such as extension, and demarcation; sharply demarcated CMs were called “geographic”) (14, 15). They collected data on general complications, lymphatic complications (lymphedema, lymphangiectasia) or venous complications (functional signs, varicose or dilated veins, leg ulcer). Additional collected data was leg hypertrophy (discrepancy of leg length and/or of girth (difference ≥ 15 mm), measured by tape, considered significant for both)) (16, 17).
Blood samples (5 ml) were collected from 113 of 120 children from the cohort, after obtaining signed consent for genetic analyses from both parents. DNA analysis was centralized in the laboratory of the Biochemistry and Molecular Biology Department of the University Hospital of Tours. Sanger sequencing was performed on DNA extracted from blood and purified by using a Qiasymphony SP system (Qiagen), then amplified by PCR with primers designed with Primer 3 software (Table SI). PCR was carried out in a volume of 20 µl, consisting of 50 ng genomic DNA, 0.15 mM of each of the 4 deoxyribonucleotides, 0.5 µM of each of the forward and reverse primers, 1X Green GoTaq Flexi Buffer (Promega Corp., Madison, WI, USA), 1.5 mM MgCl2, and 1 unit of GoTaqG2 Flexi DNA Polymerase (Promega Corp.). PCR products were sequenced in both directions with Big Dye Terminator Kit (Applied Biosystems, Foster City, CA, USA) and analysed with an ABI Prism 3130 Xl Genetic Analyser (Applied Biosystems). Investigators blinded to the patient’s clinical status (PV, AB) analysed sequences using Seqscape software (Applied Biosystems).
The primary outcome was prevalence of RASA1 variants in children with CM of the legs. In addition, the clinical phenotype of patients with a RASA1 polymorphism was described, to eventually identify correlated specific clinical signs.
Continuous variables are expressed as mean ± standard deviation (SD) and qualitative variables are reported with number (%). Fisher’s exact test was used to compare RASA1 mutations in the cohort and the general population using the Exome Variant Server (EVS) database; p < 0.01 was considered significant. To compare the characteristics between children with or without mutation, Wilcoxon rank sum tests were used for continuous variables and Fisher’s exact tests for categorical variables.
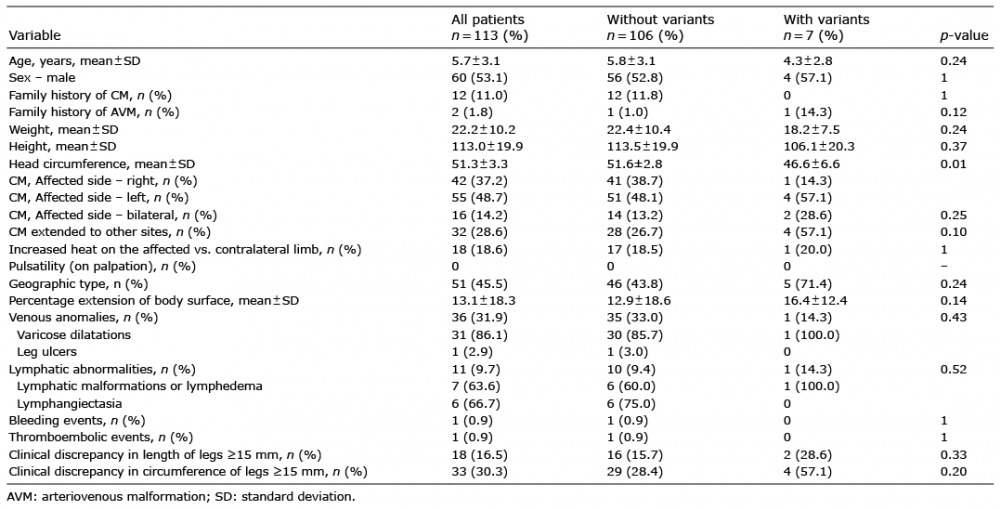
A total of 113 children were analysed for RASA1 mutations. The mean age was 5.7 ± 3.1 years, and 60 (53.1%) were male (Table I). Sixteen of the 113 patients (14.2%) had bilateral CM of the legs, and the mean body surface area of CM was 13.1 ± 18.3%. All CMs were port-wine stains (there were no cutis marmorata telangiectatica congenita, salmon patches or telangiectasia) (13); 51 patients (45.5%) had a CM of the geographic type; 32 patients (28.6%) had multi-site CMs. Venous and lymphatic abnormalities were associated in 36 (31.9%) and 11 cases (9.7%). Twelve patients (12.8%) had discrepancy in leg length and 20 (21.3%) in leg circumference.
Table I. Baseline characteristics of 113 children with capillary malformations (CM) of the legs without and with genetic RASA1 variant
Among the 113 children studied, 7 harboured heterozygous RASA1 variants (6.1%); with missense substitutions and an intronic substitution (Fig. 1, Tables I and II). None had personal medical history, morphological abnormalities, malformation syndromes, general complications, tumour development or AVM. No first-degree family history of CM was present, and none had CMs with features of those found in CM-AVM syndrome (small CMs surrounded by halo) (18).
Fig. 1. Schematic structure of the protein 120 Ras-GAP (Prosite profiles) with localization of the identified mutations (in italic). Protein domains are represented by grey boxes. SH: Src Homoly domain; PH: Pleckstrin homology domain; C2: calcium-dependant phospholipid-binding domain; GAP: GTPase activating domain.
Table II. Allelic frequencies of the RASA1 mutations identified in the cohort
Four patients carried a heterozygous p.A99V variant located in the N-terminal domain (rs111840875 [c.296c>t]). Thus, allelic frequency in our cohort was 0.018. This variant was previously recorded in the European-American population (allelic frequency 0.029). It is predicted to be neutral (score –0.342) by in silico prediction tools PROVEAN and benign (score 0.334) by Polyphen-2. The age of these 4 children was 28 months (Fig. 2a), 33 months (Fig. 2b), 35 months and 9 years (Fig. 2c). CM was unilateral for all 4 patients and extended to the whole limb for 3. The proportion of the body surface area affected by CM ranged from 7% to 40%. No CM had a geographic pattern. No case showed venous or lymphatic complications. For one patient, the lower limb with CM was hypertrophic (circumference +25 mm compared with the other limb), with no discrepancy in leg lengths. For another patient, the circumference was lower for the limb with CM than the contralateral limb (–20 mm), with no discrepancy in limb length.
Fig. 2. Examples of clinical characteristics. (a) A 28-month-old boy with widespread capillary malformations (CM) of the left leg (variant p.A99V). (b) A 33-month-old boy, showing CM extending to the back (variant p.A99V). (c) A 9-year-old girl, with CM of her right thigh (variant p.A99V). (d) A 4-year-old girl, showing a geographic-type CM of her left thigh associated with dilated veins and macrocystic lymphatic malformation of her left buttock (variant pL116V). (e) A 31-month-old girl with bilateral CMs on legs (intronic variant). (f) A 7-year-old boy showing widespread bilateral CMs (variant p.N451S).
A p.L116V variant was identified in one patient. It was considered non-pathogenic by PROVEAN and Polyphen-2 analysis. This missense variant rs149246093 (c.346c>g) was reported as very rare variant in the European-American population (0.00023). The patient was a 4-year-old girl, with a unilateral geographic-type, warm CM located on the thigh, leg and foot, spreading over 2% of the body surface, and associated with a macrocystic lymphatic malformation of the buttock. The limb affected by CM was hypertrophic, with a slight discrepancy in leg length (+12 mm), and dilated superficial veins (Fig. 2d).
A novel variant, c.539+1g>a was identified in a 31-month-old-girl. This variant is located within the donor site for splicing of intron 1. The child had bilateral asymmetric CMs, spreading over 21% of the body surface on the thighs and legs (Fig. 2e). On her right leg she had a geographic-type CM, and on the contralateral leg, a small CM on her leg and a round CM on her thigh. There was no discrepancy in the leg lengths or other abnormalities.
Finally, a variant p.N451S, located close to the Pleckstrin homology domain in the RASA1 protein was identified in a 7-year-old boy. This variation was considered non-pathogenic by PROVEAN and Polyphen-2 analysis. This missense variant, rs529871259 (c.1352a>g), was never described in the European-American population EVS, but was reported in the African population with very rare frequency (0.002). The child had CMs located on both legs, spreading over 10% of the body surface (Fig. 2f). The legs had discrepant lengths (+20 mm) but similar circumferences. The patient also had lymphedema on both feet.
This is the first study of the RASA1 gene in a population of children with CM of the legs. Among the 113 children analysed for all exons of the RASA1 gene, 7 had a heterozygous variant. All were considered non-pathogenic based on our analysis, although this was questionable for the variant N451S and for an intronic variant in intron 1.
Four children in our cohort harboured the missense variant c.296c>t, p.A99V. This variant was originally classified as a variant of uncertain significance, which was later changed to benign, as it is found in 2.9% of the European-American general population EVS. This variant was first reported by Wooderchak-Donahue et al. (8) in a patient with multifocal CMs on the head, face and chest with AVM. It was considered benign, since this patient also had another pathogenic stop codon mutation.
We observed a novel variant c. 539+1g>a at the beginning of intron 1 of RASA1, in the donor site for splicing. This variant may affect the splicing of RASA1 pre-mRNA as suggested by in silico analysis using MaxEntScan (MES; Markov model). We next searched for an impact of this variant by analysing the expression of RASA1 in RNA extracted from blood of the girl carrying the variant. We did not observe an alternative transcript by RT-PCR (data not shown). Further studies, particularly on other tissues, could be of interest.
The mutation c.1352a>g (p.N451S) was considered non-pathogenic by PROVEAN and Polyphen-2 analysis, but it is a very rare variant that was never described in the European-American population EVS, and only reported in the African population with a very rare frequency (0.002). Moreover, it is located in exon 10, where several mutations were previously associated with CM-AVM (5, 19). Further studies including protein functional tests could be interesting to attest the pathogenicity of the variant.
We did not observe missense mutations or frameshift mutations causing a premature stop codon in our study, as previously reported (6, 8, 19). The rate of RASA1 variants is lower in our cohort of children with CM on the legs than in populations with multifocal CMs: 29% and 78% in studies reported by Wooderchak-Donahue and Revencu, respectively, but those included patients with or without other vascular malformations (8, 19). Also, another study of 13 patients considered to have Klippel-Trenaunay syndrome and 17 with single CM associated with an overgrown limb found no germinal RASA1 mutations (20).
In our cohort, bilaterality of the CM tended to be more frequent (28.6% vs. 13.2%), affected the mean total percentage extension of CM to the body more (16.4% vs. 13%) and extended more often to other sites (57.1% vs. 26.7%) in children with than without RASA1 variants, although not significantly. No children with RASA1 variants had AVM or small round CMs with surrounding halo typical of those in CM-AVM syndrome. In previous reports on patients with CM and RASA1 mutations, most multifocal and bilateral CMs were associated with CM-AVM syndrome. Multifocal CMs without AVM could also be associated with RASA1 variants, but CM were more frequently patchy CMs than widespread CM extending to other sites, as observed in patients with our variants (7, 8, 20). We hypthesize that identification of a germline variation in RASA1 may favour bilateral CM compared with post-zygotic mutations that may lead to unilateral segmental CM (1). Overgrowth syndrome, described in several cases of CMs with somatic mutations of PIKCA3 (3), did not appear to be more frequent in our patients with than without RASA1 variants.
This study has a number of limitations. First, the sample size of children with RASA1 variants was too small to confirm the clinical differences with children and without variants. Secondly, the study would have been strengthened if clinical characteristics of CMs were more precisely classified (15). Thirdly, genetic analyses were performed on blood samples and yield would have possibly be higher with lesional tissue analysis. Also, we focused our search on germline mutations of the RASA1 gene; 1 case of CM-AVM with mutation on EBH4 has been reported recently (21). Finally, the conditions involving CMs were heterogeneous, and no disease-causing relationship could be proven in these cases.
In conclusion, variants of RASA1 are rarely found with CM of the legs without associated CM-AVM syndrome. We found no clinical phenotype associated to germline RASA1 mutation. However, mutations in the promoter of RASA1 could be investigated in patients with CM. Similarly, deletions or duplications of particular exons or the entire RASA1 gene could be investigated. Use of comparative genetic hybridization array or next-generation sequencing studies of cohorts of patients with CM could lead to more discovery of RASA1 mutations in this population.
The authors are indebted to Mrs A. Darmaillacq, from CHRU Tours, and Mrs M. Carriot, CIC Tours, France, for their technical help.
ClinicalTrials.gov registration: NCT01364857.
Funding sources: the study was funded by the French Ministry of Social Affairs and Health (French National Program of Clinical Research (PHRC-N), 2009)
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize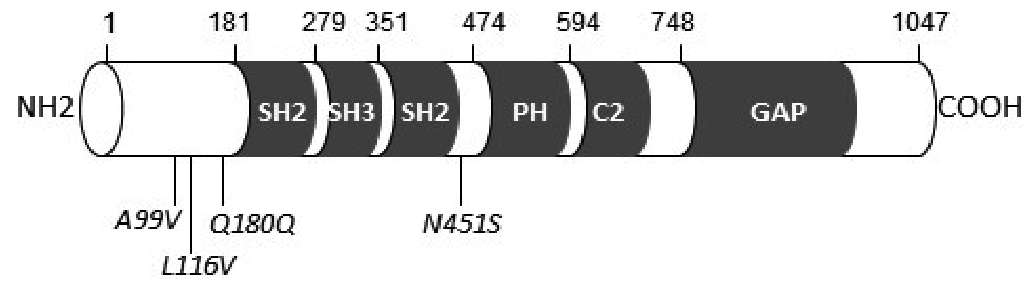 Click to show fullsize
Click to show fullsize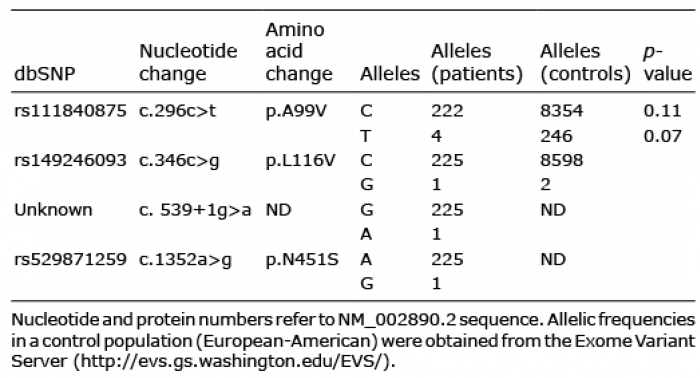 Click to show fullsize
Click to show fullsize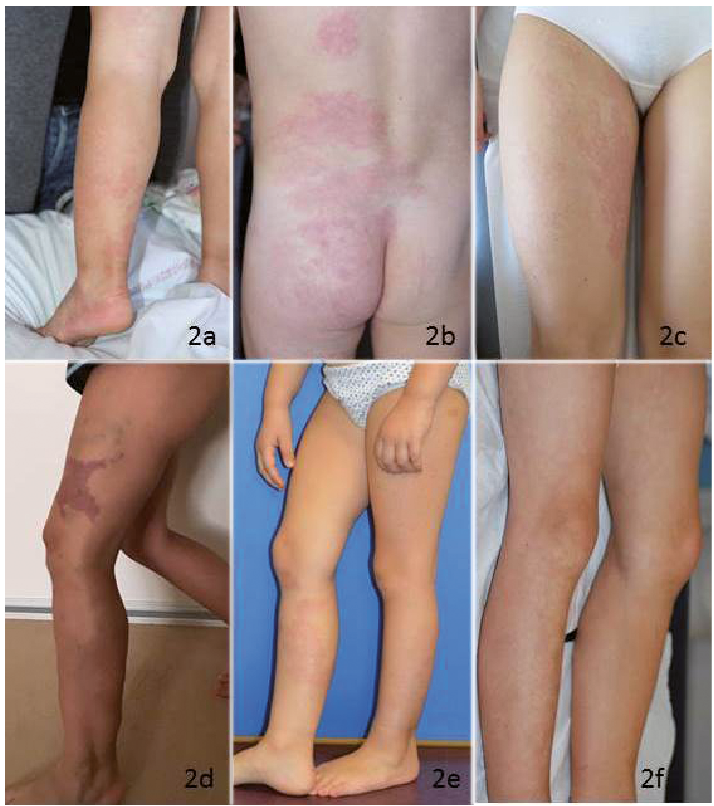 Click to show fullsize
Click to show fullsize