


Department of Dermatology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45, Yushima, Bunkyo-ku, 113-8519, Tokyo, Japan. *E-mail: hashderm@tmd.ac.jp
Accepted Jan 8, 2018; Epub ahead of print Jan 9, 2018
Adult-onset Still’s disease (AOSD) is a systemic inflammatory condition featuring a high spiking fever, arthralgia, leukocytosis with neutrophilia, and skin rash. The typical skin rash in AOSD is an evanescent, salmon-pink erythema that mainly involves the extremities (1). While various skin symptoms are reported as atypical skin rashes in AOSD (1–3), few reports mention generalized purpura (1, 4).
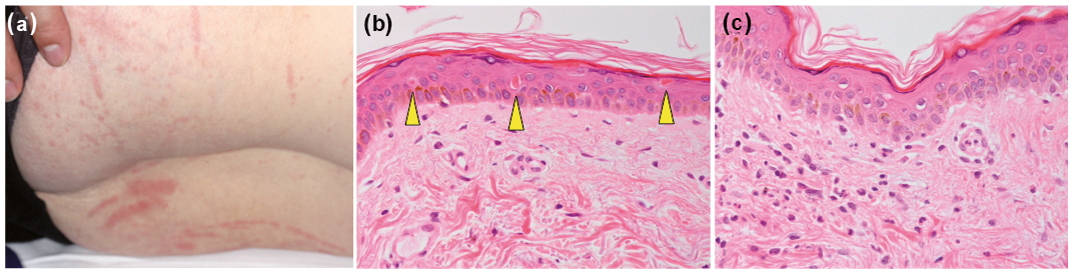
A 65-year-old Japanese woman presented with a 2-week history of pyrexia, arthralgia, sore throat and skin rash. Ten years previously she had been diagnosed with Behçet’s disease (BD) due to the presence of recurrent oral aphthous ulcers and erythema nodosum-like skin lesions. She had been treated with colchicine (1.5 mg/day) and tacrolimus (1 mg/day) and had been in remission. Physical examination revealed multiple, non-palpable, erythematous macules (5–10 mm in diameter) as linear stripes on her trunk and limbs (Fig. 1a). These macules were obvious when she had a fever, but disappeared in the absence of fever. Bilateral swollen tonsils were noted. Laboratory data revealed a high white blood cell count (16,100/µl with 81% neutrophils), elevated C-reactive protein levels (CRP 17.09 mg/dl; reference < 0.3 mg/dl) and serum hepatic/biliary enzyme levels (aspartate transaminase, 84 IU/l; alanine transaminase 110 IU/l; γ-glutamyltransferase, 105 IU/l (reference ≤ 35 IU/l); lactate dehydrogenase 615 U/l (reference 124–222 U/l)) and normal bilirubin levels (1.0 mg/dl). Serum ferritin levels were elevated (3,380 ng/ml; reference 3–120 ng/ml). Serum immunoglobulin, IgD, IgG, IgA, and IgM, levels were normal. Negative results were obtained for the presence of antinuclear antibodies and rheumatoid factor. Serum interleukin (IL)-1β (2.87 pg/ml; reference < 0.928 pg/ml), IL-6 (121 pg/ml; reference < 2.41 pg/ml), and IL-18 (1,250,000 pg/ml; reference < 211 pg/ml) levels were elevated. The patient expressed both human leukocyte antigen (HLA)-B51 and HLA-B54. Urinalysis was normal. Blood bacterial cultures yielded negative results. Whole-body computed tomography revealed splenomegaly, but no lymphadenopathies, infection foci, or malignant tumours. Ophthalmological examination and echocardiogram were both normal. A skin biopsy obtained from a macule showed dyskeratotic cells in all levels of the epidermis, vacuolar degeneration in the epidermis (Fig. 1b), and slight perivascular and interstitial lymphoneutrophilic infiltrates in the upper dermis (Fig. 1c). Neither vasculitis nor periadnexal infiltrate was found. Therefore, a diagnosis of AOSD was made. Intravenous pulse methylprednisolone therapy (1 g/day for 3 days) and oral corticosteroids (40 mg/day) were administered; a switch from tacrolimus to azathioprine (50 mg/day) was also made. Her symptoms partially improved. However, 1 day after the completion of methylprednisolone therapy, when the peripheral platelet count was normal, multiple, slightly-palpable purpura (approximately 5 mm in diameter) appeared on the trunk (Fig. 2a, b) and disappeared spontaneously within 3 days. When serum ferritin and CRP levels increased again 2 weeks later, generalized purpura recurred on the trunk and proximal limbs. Negative results were obtained for the presence of anti-neutrophil cytoplasmic antibodies. A bone marrow biopsy was negative for haemophagocytic syndrome. A second skin biopsy of the purpura revealed scattered dyskeratotic cells and vacuolar degeneration in the epidermis with erythrodiapedes and perivascular lymphocytic infiltrates in the upper dermis (Fig. 2c). Neither vasculitis nor periadnexal infiltrate was detected. AOSD relapse was considered, and steroid therapy was restarted, with subsequent improvement in symptoms.
Fig. 1. Clinical and histopathological features at initial presentation. (a) Non-palpable, erythematous papules and macules (5–10 mm in diameter) were obvious as linear stripes on the proximal limbs, suggestive of Koebner phenomenon. (b and c) Histopathological findings of erythematous macules. (b) Dyskeratotic cells (yellow arrowheads) in all levels of the epidermis and (c) vacuolar degeneration in the epidermis were accompanied by perivascular and interstitial lymphoneutrophilic infiltrates in the upper dermis. No vasculitis was seen (haematoxylin and eosin staining; original magnification ×200).
Fig. 2. Clinical and histopathological features of generalized purpura. (a, b) Multiple, slightly-palpable purpura (approximately 5 mm in diameter) were scattered on the trunk. (b) Each purpuric rash barely coalesced. This skin symptom accompanied adult-onset Still’s disease (AOSD). (c) Histological findings of purpura. Dyskeratotic cells (yellow arrowheads) and vacuolar degeneration in the epidermis were accompanied by erythrodiapedes and perivascular lymphocytic infiltrates in the upper dermis. No vasculitis was seen (haematoxylin and eosin staining; original magnification ×100).
The patient’s initial skin eruptions featured a linear configuration suggestive for Koebner phenomenon. Histopathological examination showed scattered dyskeratotic cells in the epidermis. These clinical and pathological findings are characteristic of atypical skin manifestations in AOSD, termed “persistent pruritic papules and plaques” or “flagellate erythema-like eruption” (2, 3). Sweet’s syndrome (SS) should also be considered, since Koebner phenomenon and/or salmon urticarial rash can sometimes be seen in SS (5) and the patient expressed HLA-B54, which is related to SS. However, SS is questionable because scattered dyskeratotic cells in the epidermis are not seen in SS and because of the lack of massive neutrophilic infiltrate in the upper dermis seen in SS.
Later, generalized purpura appeared twice, accompanied by AOSD. BD-induced purpura is questionable because no other BD symptoms were apparent. In addition, histological examination revealed scattered dyskeratotic cells in the epidermis, which are characteristic of AOSD, but not of BD. Interface dermatitis was also seen in the patient, but no periadnexal infiltrate. This finding is also compatible with AOSD (1–3). Drug-induced purpura is also questionable because the purpura resolved, even during drug treatment. Therefore, the generalized purpura in this patient was associated with AOSD. The mechanism responsible for the generalized purpura is unclear because the patient had no evidence of thrombocytopaenia, coagulopathy, or vasculitis.
Interestingly, the patient had been diagnosed with BD before onset of AOSD. To our knowledge, the coexistence of AOSD and BD has not been reported, whereas the coexistence of AOSD and SS has been reported previously (6). AOSD, BD and SS have a similar pathogenesis, wherein neutrophilic hyperactivity/dysfunction plays a pathogenic role (7, 8). Notably, the patient expressed HLA-B51 and HLA-B54, which are related to BD (8) and SS (9), respectively. In addition, the patient had a high level of serum IL-18, which is known to play a significant role in the pathogenesis of BD (10) and AOSD (11). This genetic background, accompanied by the overproduction of IL-18, may contribute, at least partially, to the coexistence of AOSD and BD in this patient and to the occurrence of generalized purpura as a rare skin manifestation in cases of AOSD.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize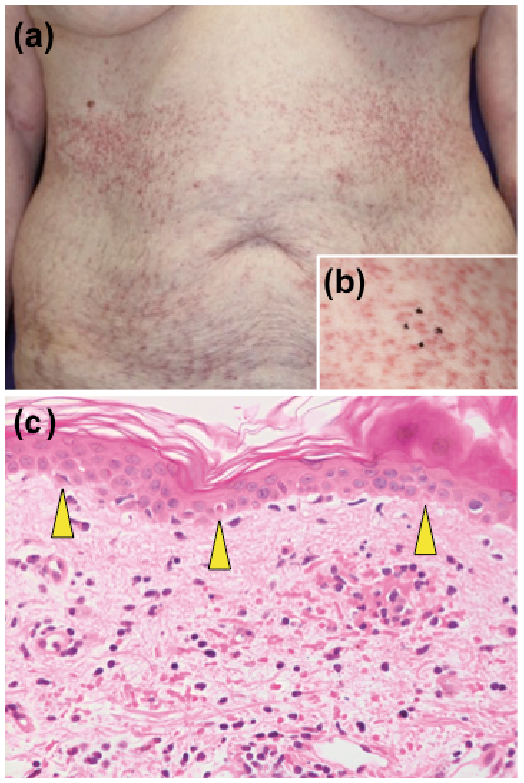 Click to show fullsize
Click to show fullsize