


Department of Dermatology, Lauriston Building, Lauriston Place, Edinburgh, EH3 9HA, Scotland. E-mail: dominic.tabor@nhslothian.scot.nhs.uk
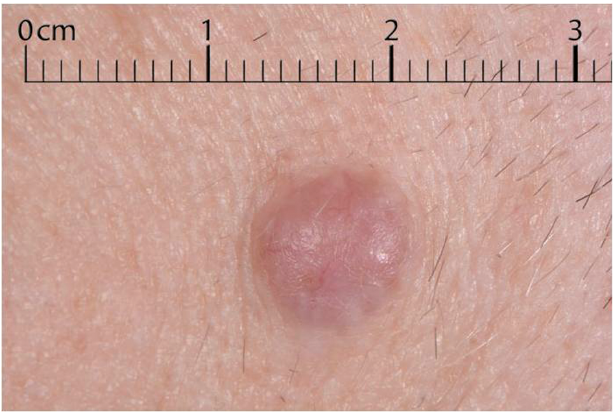
A 65-year-old man presented with an 8-year history of a slowly enlarging, asymptomatic nodule on his left temple (Fig. 1), which was erythematous and telangiectatic on examination, but otherwise bland dermoscopically. He was systemically well, but had a background of schizophrenia and type 2 diabetes mellitus. Full skin survey and computed tomography of the chest, abdomen and pelvis did not reveal any other abnormalities.
Histological examination following elliptical excision of the nodule with a 4 mm margin demonstrated a well-demarcated nodular infiltrate of lymphoid aggregates with reactive germinal centres, with numerous plasma cells and histiocytes present (Fig. 2).
What is your diagnosis? See next page for answer.
Fig. 1. Asymptomatic nodule on the temple.
Fig. 2. Histology of excised nodule (H&E x40).
Acta Derm Venereol
Diagnosis: Cutaneous Rosai-Dorfman disease (RDD)
Histological staining revealed that the histiocytes stained positive for CD68 and CD168, with a subpopulation of multinucleate histiocytes staining positive for S100. Some histiocytes were large and multinucleated, with emperipolesis noted (Fig. 3).
These clinicopathological features, in the absence of lymphadenopathy or other systemic features, were in keeping with a diagnosis of cutaneous RDD.
RDD is a rare, benign histiocytosis, which was first described in 1965 (1) and recognised as a distinct clinical entity in 1969 (2), when it was termed sinus histiocytosis with massive lymphadenopathy. The classical presentation is of massive cervical lymphadenopathy, with fevers, weight loss, a raised ESR, and a polyclonal gammopathy often noted. Extranodal disease occurs in approximately 40% of cases, with skin the most common site (3).
Cutaneous RDD, where skin lesions are found without lymphatic or other tissue involvement, was first described in 1978 (4) and is rarer than the systemic form. The epidemiology differs between the two, with cutaneous RDD more common in women (F:M 2:1), older patients (mean age 43), and in patients of white or East Asian descent (5). Although long-term follow-up in the literature is limited, purely cutaneous RDD does not appear to develop systemic involvement. It may therefore be regarded as a distinct clinical entity from systemic RDD, although it is not possible to distinguish between cutaneous and systemic RDD on the basis of the clinical or histological findings from a cutaneous lesion.
The clinical appearances of cutaneous RDD are variable, with nodules, papules, and plaques all described, most commonly red in colour. Histologically, a mixed infiltrate of plasma cells and large, polygonal histiocytes with clear cytoplasm is seen. Histiocytes demonstrate emperipolesis, which describes the presence of intact lymphocytes, plasma cells, or neutrophils within the histiocyte cytoplasm. This differs from phagocytosis in that the cells are not destroyed. The histiocytes lack Birbeck granules, and stain positively for S100 and sometimes CD68, but are negative for CD1a. The S100 positivity and presence of emperipolesis helps to differentiate RDD from differentials, which include Langerhans cell histiocytosis, malignant histiocytosis, large cell or Hodgkin’s lymphoma, and melanoma. Plasma cells are often also seen. The aetiology is unclear, but autoimmune disease, haematological malignancy, or viral infection, particularly with Epstein-Barr virus, have been suggested as potential triggers. Spontaneous resolution has been reported (5). Multiple treatments for persistent cutaneous RDD have been reported with variable success, including surgical excision, systemic or intralesional steroids, retinoids, thalidomide, cryotherapy, radiotherapy, imatinib, and dapsone. Our patient remains under regular review.
Fig. 3. S100 x100 demonstrating emperipolesis.


 Click to show fullsize
Click to show fullsize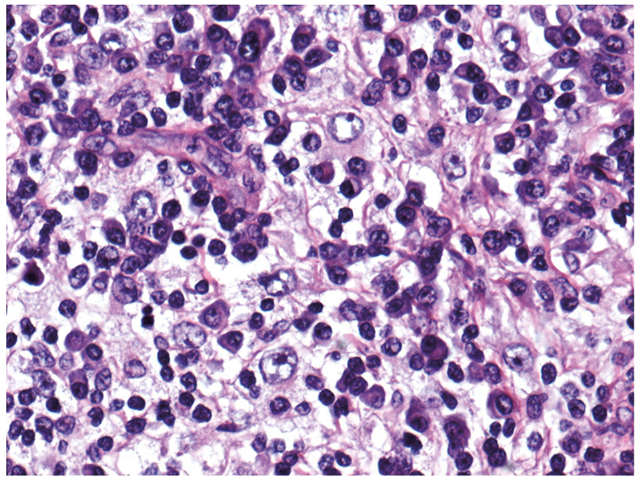 Click to show fullsize
Click to show fullsize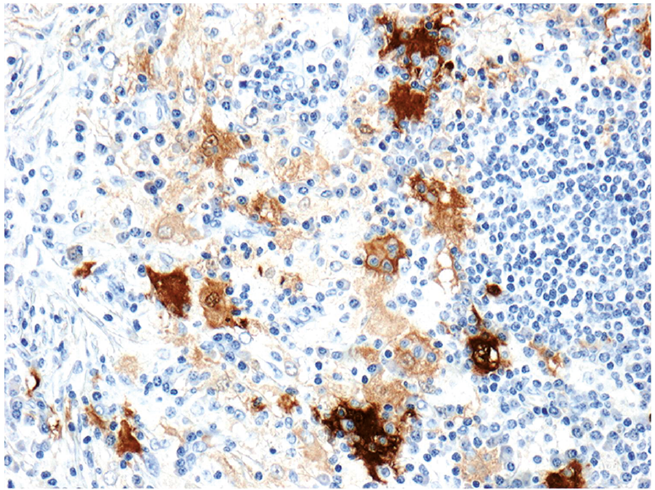 Click to show fullsize
Click to show fullsize