


1Department of Dermatology, Venerology and Allergology, University Medical Center, Ruprecht-Karls-University, Im Neuenheimer Feld 440, DE-69120 Heidelberg, and 2Dermatopathology Friedrichshafen, Friedrichshafen, Germany. E-mail: julia.winkler@med.uni-heidelberg.de
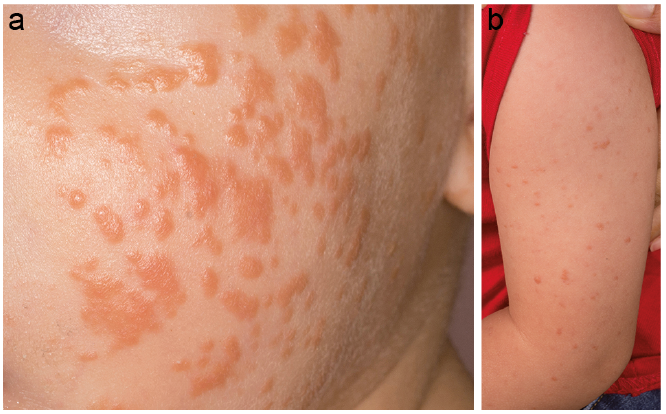
An otherwise healthy 10-month-old male infant presented with a 2-month history of slightly itchy yellow-brownish papules, which were most evident on his cheeks (Fig. 1a). Examination revealed further papules on his arms (Fig. 1b) and trunk. His family history was inconspicuous. Blood tests, including full blood count, liver-function, kidney-function and C-reactive protein level, were within normal limits. Hepatitis B and Epstein–Barr virus (EBV) serology were negative. Topical steroid and antibiotic therapy had no clinical benefit. Due to persistent papules a lesion on the arm was biopsied 2 months later. Histopathology revealed a dermal infiltrate of non-epidermotropic histiocytes admixed with some lymphocytes (Fig. 2a). Histiocytes stained positive for CD68 (Fig. 2b) and S100 (Fig. 2c) and lacked expression of CD1a and langerin (CD207). Proliferative activity (Ki67) was low.
What is your diagnosis? See next page for answer.
Fig. 1. (a) Multiple yellow-brownish papules on the cheeks. (b) Papules distributed on the arms and single papules localized on the trunk.
Fig. 2. (a) Section of a cutaneous papule from the upper arm revealing orthokeratosis, a normal epithelium and many dermal histiocytes admixed with some lymphocytes (haematoxylin-eosin stain, original magnification ×100). Immunohistochemical stain for: (b) CD68 and (c) S100 (original magnification ×100).
Acta Derm Venereol
Diagnosis: Benign cephalic histiocytosis
Clinic-pathological correlation was consistent with a diagnosis of benign cephalic histiocytosis (BCH), a rare non-Langerhans cell histiocytosis that typically affects otherwise healthy infants. It was first described by Gianotti et al. in 1971 (1). Sixty cases have been reported to date (2, 3). However, it has been suggested that BCH is under-recognized (2). Clinical differential diagnoses include plane warts, Spitz naevi and urticaria pigmentosa, while histopathological differential diagnoses comprise juvenile xanthogranulomatosis (JXG), generalized eruptive histiocytosis (GEH) and sarcoidosis. Making a precise diagnosis based on clinical appearance and histopathology is crucial in order to determine the therapeutic procedure and prognosis. In most cases BCH presents with prominent facial papules, being the first skin manifestation. However, involvement of extrafacial skin was described in most of the infants (2). Various entities of non-Langerhans cell histiocytosis present a spectrum of disease with overlapping characteristics (4). Two cases of BCH with transformation into JXG have been reported (5, 6). Moreover, cases with overlapping characteristics of JXG and BCH have been described (7). It has been discussed whether BCH represents a localized variant of GEH (4, 8). In a blinded histological study BCH, GEH and early non-xanthomatous JXG did not show significant differences (9). BCH may be differentiated from JXG by the absence of foamy cells and Touton giant cells (10). In contrast, Langerhans cell histiocytoses show reniform nuclei, eosinophil cytoplasm and epidermotropism, which were absent in our patient. Ultrastructural features of BCH include comma-shaped intracytoplasmatic bodies and coated vesicles in the histiocytes (6, 11, 12). Immunohistochemistry is essential for further characterization and differentiation. While Langerhans cell histiocytoses are characterized by expression of S100, CD1a and langerin (4), S100 staining is usually negative in BCH. However, as in our patient, some cases with weak expression of S100 were reported (4, 13).
Altogether, the clinician needs to take into account various factors, such as distribution of skin lesions, age of onset, associated symptoms and histopathological features in order to make a diagnosis. Our patient was referred to the children’s hospital and an ophthalmologist. Paediatric examination did not result in any pathological findings. Blood count did not reveal any atypical lymphocytes. During the nearly 5-month clinical follow-up skin manifestations persisted with a slight increase of the number of lesions on the arms. BCH has a quite favourable prognosis usually showing spontaneous regression at a median time of 50 months (3, 4). Lesions may leave hyperpigmentation or scars (7, 12). Apart from one case of diabetes insipidus and another child who developed diabetes mellitus, systemic involvement has not been reported in BCH (14, 15). In the current case we did not recommend any further local or systemic therapy.
In conclusion, dermatologists and paediatricians should be familiar with this uncommon entity and its differential diagnoses in order to recommend the correct therapeutic approach.


 Click to show fullsize
Click to show fullsize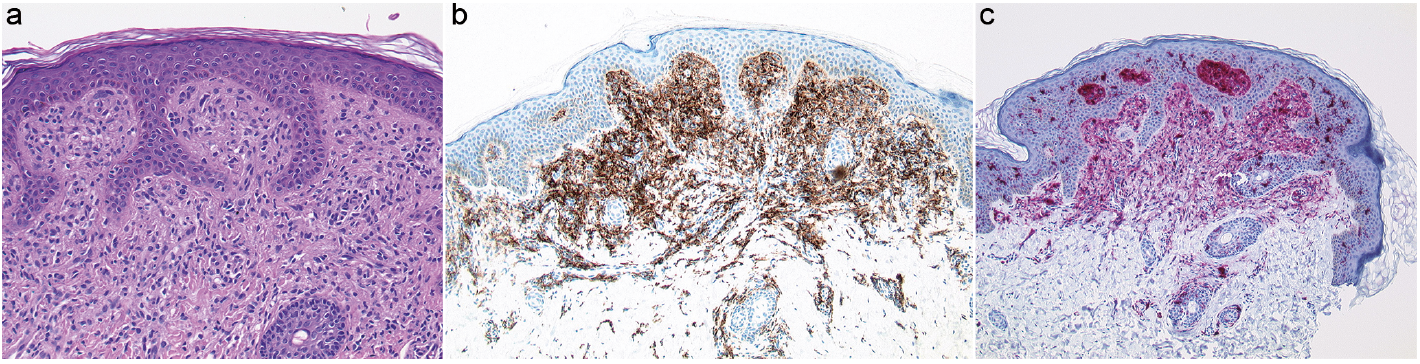 Click to show fullsize
Click to show fullsize