


Departments of 1Dermatology & Venereology, 4Pediatrics, 5Anatomy and 6Pathology, All India Institute of Medical Sciences, 2CSIR-Institute of Genomics & Integrative Biology, New Delhi, and 3Academy of Scientific and Innovative Research, Council of Scientific and Industrial Research (CSIR), New Delhi, India
Recent advances in the field of genomics have seen the successful implementation of whole exome sequencing as a rapid and efficient diagnostic strategy in several genodermatoses. The aim of this study was to explore the potential of molecular studies in dystrophic epidermolysis bullosa (DEB) in India. Whole exome sequencing was performed using genomic DNA from each case of epidermolysis bullosa, followed by massively parallel sequencing. Resulting reads were mapped to the human reference genome hg19. Sanger sequencing subsequently confirmed the potentially pathogenic mutations. Whole exome sequencing of 18 patients with DEB from 17 unrelated Indian families revealed 20 distinct sequence variants in the COL7A1 gene including 2 widely prevalent mutations. Dominant inheritance was seen in 7 patients, while 11 patients showed a highly variable recessive DEB. This preliminary study using exome sequencing is clearly encouraging and will serve as the basis for future large-scale molecular studies to actively identify and understand DEB in the Indian population.
Key words: dystrophic EB; collagen VII; whole exome sequencing.
Accepted Mar 27, 2018; Epub ahead of print Mar 27, 2018
Acta Derm Venereol 2018; XX: XX–XX.
Corr: Gomathy Sethuraman, Department of Dermatology, All India Institute of Medical Sciences, New Delhi-110029, India. E-mail: kgsethu@yahoo.com/aiimsgsr@gmail.com and Sridhar Sivasubbu, CSIR-Institute of Genomics & Integrative Biology, Mathura Road, New Delhi, India. E-mail: sridhar@igib.in
Dystrophic epidermolysis bullosa is a rare and devastating genetic skin disease associated with many complications including skin cancer. In the absence of a cure, making an accurate genetic diagnosis is therapeutic in its own right to the patients and their families as it potentially benefits them in terms of management and counseling. To this extent, this study provides an insight into the mutational spectrum of a small group of Indian patients with dystrophic epidermolysis bullosa and also stresses the advantage of next generation DNA sequencing technologies in aiding the accurate diagnosis of patients with epidermolysis bullosa.
Dystrophic epidermolysis bullosa (DEB) is a rare genetic skin disease characterized by trauma-induced muco-cutaneous blistering associated with scarring, milia and nail dystrophy. Depending on the mode of inheritance, DEB is categorized into dominant (DDEB) and recessive (RDEB) forms that are further divided into many sub-types based on clinical presentation. All sub-types are associated with mutations in the COL7A1 gene encoding type VII collagen, a major stabilizing molecule of the dermoepidermal junction (DEJ) (1).
Despite well-characterized genetic studies from different ethnic backgrounds, identifying several recurrent and region-specific mutations, molecular diagnosis of DEB is still challenging. This is because most patients harbour mutations that tend to be family specific, as evident from the large repertoire of variations identified across the entire COL7A1 gene, spanning 118 exons. To make it more complex, wide phenotypic heterogeneity is also observed across the different sub-types of DEB, with multiple mutations resulting in a similar phenotype, and the same mutation showing variable clinical presentations, in addition to considerable inter- and intra-familial variations (2, 3).
Prior to the advent of next-generation DNA sequencing (NGS) technologies, molecular diagnosis of DEB was based on traditional Sanger sequencing of either the hot-spot regions or the entire COL7A1 gene, requiring more than 70 primer sets; a process that is tedious, time consuming and expensive (4). Due to its high throughput nature and ability to simultaneously sequence multiple genes, NGS, especially whole exome sequencing (WES), has revolutionized the molecular approach and provided a fast and efficient diagnostic strategy in several genodermatoses (4, 5). In view of the scarcity of, and need for, molecular studies for DEB in India, the aim of this study was to explore the potential of WES in the phenotype and genotype characterization of patients with DEB, as part of a larger effort in understanding the mutational spectrum of patients with EB in India.
In this study, 18 consecutive patients (PT01–PT18) from 17 unrelated Indian families, clinically diagnosed with DEB, were recruited at a single tertiary care centre in New Delhi between July 2013 and December 2015. The cohort included 12 boys and 6 girls. Age of presentation ranged from 2 months to 24 years. After obtaining informed consent, detailed clinical history, cutaneous and systemic examination was performed. Baseline clinical photographs were recorded in all patients. The Institutional Ethics Committee approved the study (IEC number IESC/T-147/04.04.2014).
A 3-mm skin punch biopsy was taken from normal-appearing skin, usually on the thigh, in all affected patients, as recommended (6), and was subjected to immunofluorescence antigen mapping (IFM) using a limited panel of commercially available monoclonal antibodies that included keratin 14 (NCL-LL002 (1:50), Novocastra, Newcastle, UK); type IV collagen (CIV-22 (1:100), Dako, Carpentaria, CA, USA), type VII collagen (NCL-COLL-VII (1:50), Novocastra, Newcastle, UK) and laminin-332 (P3H9-2 (1:50), AbCam, Cambridge, UK).
Genomic DNA was isolated using the salt extraction method from fresh peripheral blood samples collected from all affected patients and their parents, where available. Whole-exome capture sequencing was performed as described elsewhere (7, 8) using Nextera Rapid Capture (61 MB, version 4, Illumina, San Diego, CA, USA) followed by massive parallel sequencing on Illumina Hiseq 2500 platform (Illumina, San Diego, CA, USA). Resultant reads were mapped to human reference genome (hg19). Variant calling was performed using Genome Analysis Tool Kit (GATK) pipeline (9) in standard vcf format. The resultant variant calls were annotated using ANNOVAR tool (10) against publically available databases and analysed via SIFT (http://sift.jcvi.org/; pathogenic if score ≤ 0.05) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/; pathogenic if score ≥ 0.447) for its pathogenicity. Sanger Sequencing (Big Dye Terminator Cycle Sequencing kit v3.1, Applied Biosystems, Foster City, USA) was used to validate the candidate variants in affected patients and their segregation in families.
Overall, dominant inheritance was observed in 7 patients (PT01–PT07) and recessive inheritance in 11 patients (PT08–PT18). All families had at least one affected child, except family 4, which had 2 affected siblings (PT04 and PT05). History of consanguinity was recorded in only 3 families (PT07, PT09 and PT17).
Clinical characteristics. All affected individuals presented with disease onset from the first week of life, except PT01 and PT03, who had trauma-induced blisters or erosions since 3 months and 5 years of age, respectively. All patients had relatively mild disease limited to elbows, knees and ankles, except PT06 and PT07, who had generalized blisters predominantly over the lower extremities. None of them reported having pruritus. Other disease features included mild atrophic scarring and milia formation with occasional erosions in the oral mucosa. Nail involvement was variable, in that all patients showed toenail dystrophy, while only a few had both toenail and fingernail dystrophy (PT03, PT05, PT06 and PT07). All probands were born to non-consanguineous parents with significant family history in 5 patients (PT01, PT03, PT04, PT05 and PT07). The fathers of PT01 and PT03 and mothers of PT04, PT05 and PT07 presented with nail dystrophy only and recalled the development of blisters, limited to knees and elbows, in early childhood that had improved until complete resolution as adults.
Immunofluorescence antigen mapping findings. Skin biopsies showed only focal cytolysis at the DEJ in 4 patients (PT02, PT04, PT06 and PT07) and no split in 3 patients (PT01, PT03 and PT05). IFM analysis showed a mild to moderate (+ or ++) positive staining for COLVII antibody in all patients compared with a normal healthy control (+++), consistent with a diagnosis of DDEB.
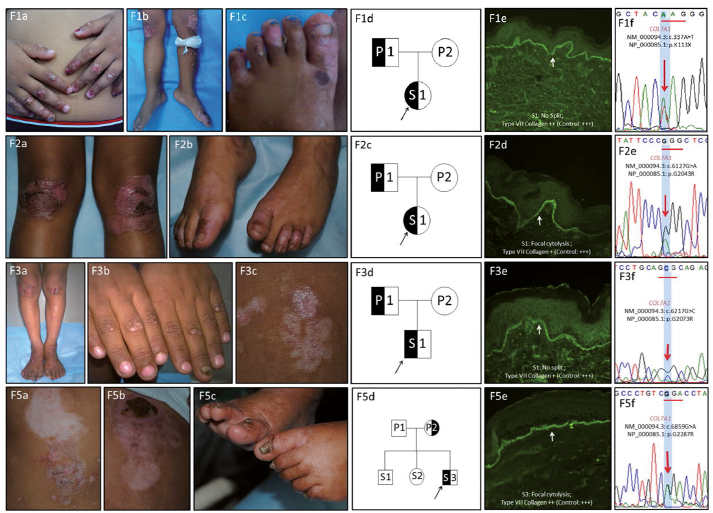
Molecular findings. Five distinct heterozygous sequence variants were identified that resulted in single base substitutions in COL7A1 (NM_000094.3; NP_000085.1) gene: 2 missense glycine substitutions that have been previously reported in DDEB patients and 3 novel variations that included 2 glycine substitutions at previously reported nucleotide positions in PT03, PT04, PT05 (Exon 75: c.6217G>C; p.Gly2073Arg) and PT07 (Exon 58: c.5201delG; p.Gly1734fs) and a nonsense variation in PT01 involving the substitution of a lysine residue with a stop codon (Exon 3: c.337A>T; p.Lys113X). Among the previously reported variations, both PT02 (Exon 73: c.6127G>A; p.Gly2043Arg) and PT06 (Exon 87: c.6859G>A; p.Gly2287Arg) showed the presence of widely prevalent hot-spot mutations. Sanger sequencing confirmed the pathogenic variations in the affected individuals, along with maternal inheritance in PT04, PT05 and PT07 and paternal inheritance in PT01 and PT03, respectively. Blood samples of the parents of PT02 and PT06 were unavailable for segregation analysis. How-ever, they were clinically unaffected and apparently healthy, hinting at a de novo nature of the mutations. Fig. 1 and Tables I and II summarize the phenotype and genotype characteristics of patients with DDEB.
Fig. 1. Phenotype and genotype spectrum of dominant dystrophic epidermolysis bullosa (DDEB) patients in India. Clinical findings of corresponding DDEB families: F01, 03, 05 (a–c) and F02 (a, b): mild blisters localized to bony prominences along with mild nail dystrophy and milia formation. Pedigree charts of corresponding families (01d, 02c, 03d, 05d): Symbols: half-black=affected patients; open symbols=unaffected individuals. Immunofluorescence antigen mapping (IFM) findings for type VII collagen (clone: CIV-22, Dako Carpentaria, CA, USA): skin biopsy sections showing focal cytolysis (2d and 5e) or no split (1e and 3e) along with reduced staining (+) for type VII collagen (2d and 3e) and near normal (++) staining (01e and 05e) compared with normal healthy control (+++). Sequence chromatograms (01f, 02e, 03f and 05f) of corresponding patients depicting the mutations in COL7A1 gene.
Table I. Phenotypic spectrum of patients with dominant dystrophic epidermolysis bullosa from India
Table II. Genotype spectrum of patients with dominant dystrophic epidermolysis bullosa from India
Clinical characteristics. Presentation was highly variable with a generalized severe (RDEB_GS) phenotype in 5 patients (PT08–PT12) and a milder, generalized intermediate (RDEB_GI) phenotype in the remaining patients (PT13–PT18).
All patients, except PT18, had extreme skin fragility soon after birth that resulted in extensive scarring and atrophy along with milia formation. In 2 patients (PT16 and PT17), the disease severity reduced with age in spite of developing frequent new blisters. Nail involvement was significant with complete loss of all 20 nails in 6 patients (PT08–PT09, PT13–PT15 and PT17) and nail dystrophy with loss of some finger and toenails in the remaining 5 patients (PT10–PT12, PT16 and PT18). Mucosal involvement was severe with repeated oral cavity blisters, dysphagia due to oesophageal strictures, recurrent episodes of constipation, bleeding per rectum and severe dental caries. As a result, severe nutritional compromise was noted in most patients.
In 4 patients (PT08, PT09, PT13 and PT15), opening of mouth was restricted and tongue has become less mobile. Scarring alopecia was noted in 3 patients (PT08, PT13 and PT15) while cicatricial symblepharon (PT08) and corneal opacities (PT12) were noted in one patient each. Although, none of the patients developed mitten deformities at the time of presentation, partial inter-digital fusion and webbing was observed in 4 patients (PT08–PT10 and PT15). Flexion contractures, as a result of repeated scarring and blistering were noted in 2 patients (PT10 and PT13).
All affected individuals were born to non-consanguineous families, except PT09 and PT17. Significant family history was noted in only one patient (PT08), where the elder sibling died at 11 months of age with severe disease. None of the patients showed any signs of squamous cell carcinoma at the time of presentation.
IFM findings. In this subset, the plane of cleavage at the DEJ was variable with a clear or partial sub-epidermal split in 8 patients (PT08–PT15) and focal cytolysis in 3 patients (PT16–PT18), respectively. In 5 cases, IFM analysis showed completely absent staining COLVII antibody while the remaining 6 cases showed mild to moderate (+ or ++) staining compared with a normal healthy control (+++), reflecting the presence of relative amounts of functional type VII collagen at the DEJ.
Molecular findings. Molecular characterization of this group was heterogeneous with homozygous sequence variants in 5 patients (PT08–PT11 and PT13) and compound heterozygous inheritance in 6 patients (PT12, PT14–PT18). Among the homozygous variants, 5 distinct (2 novel and 3 previously reported) mutations were identified involving 2 nonsense variations in PT08 (Exon 2: c.172dupC; p.R58fs) and PT09 (Exon 14: c.1893G>A; p.W631X), 2 splice-site mutations in PT10 (Intron31–Exon32: c.3832-2A>G) and PT11 (Intron109–Exon110: c.8047-1G>A) and a missense glycine substitution in PT13 (Exon73: c.6073G>A; p.Gly2025Ser).
Among the patients with compound heterozygous inheritance, a total of 3 nonsense, 4 missense, 1 splice-site and 2 frameshift mutations were identified, of which 5 were reported previously and 5 were novel variations. Of these, only 4 were glycine substitutions. Two mutations were common between 2 unrelated families each (PT12/PT17 and PT14/PT18). In all the affected patients, none of the parents who were heterozygous for the corresponding mutations had any muco-cutaneous or nail abnormalities. Figs 2 and 3 and Table SI and Table SII summarize the phenotype and genotype characteristics of the patients with RDEB.
Fig. 2. Phenotype and genotype spectrum of recessive dystrophic epidermolysis bullosa (RDEB)_generalized severe (GS) patients in India. Clinical findings of corresponding RDEB-GS families: F07-09 (a–c): GS scarring blisters with milia formation. Severe nail involvement is seen with complete loss of 20 nails in some patients. Pedigree charts of corresponding families (07-09d). Symbols: full-black=affected patients; open symbols=unaffected individuals. Immunofluorescence antigen mapping (IFM) findings for type VII collagen (clone: CIV-22, Dako Carpentaria, CA, USA): skin biopsy sections showing a sub-epidermal split with absent staining for type VII collagen (07-09e) compared with normal healthy control (+++). Sequence chromatograms (07-09f) of corresponding patients depicting the homozygous mutations in the COL7A1 gene.
Fig. 3. Phenotype and genotype spectrum of recessive dystrophic epidermolysis bullosa (RDEB)_generalized intermediate (GI) patients in India. Clinical findings of corresponding RDEB-GI families: generalized severe scarring blisters with milia formation. Severe nail involvement is seen with complete loss of 20 nails in some patients. Flexion contracture deformities are seen in F12 (F12c), while interdigital fusion of toes is seen in F14c. Pedigree charts of corresponding families (12e and 15e; 13d and 14d). Symbols: full-black- homozygously affected patients; half pattern-half black-heterozygously affected patients; half pattern-healthy carriers and open symbols-unaffected individuals. Immunofluorescence antigen mapping (IFM) findings for type VII collagen (clone: CIV-22, Dako Carpentaria, CA, USA). Skin biopsy sections showing a sub-epidermal split with absent staining (13e and 14e) and reduced staining (+) (12f and 15f) for type VII collagen compared with a normal heathy control (+++). Sequence chromatograms of corresponding patients depicting the compound heterozygous mutations in the COL7A1 gene.
DEB is perhaps the most frequently studied sub-type of EB and is inherited in both autosomal dominant and recessive forms (1). Although particular genotype and phenotype characteristics of DEB have been elucidated through extensive molecular studies in several countries, little is known about this group in the Indian population. The present study in a small cohort of 18 Indian patients with DEB is aimed at improving this understanding. Our results largely comply with the widely accepted genotype-phenotype correlation concepts.
DDEB is usually associated with glycine substitutions in the triple helix domain of the COL7A1 gene, with a majority of them residing in exons 73–75 (2, 3). Among the 7 DDEB patients in our cohort, 6 patients showed glycine substitution mutations, with 4 (66.7%) of them in this hot-spot region. Identification of p.2043G>R in PT02 and p.2287G>R in PT06 reiterates the fact that these are widely prevalent COL7A1 mutations implicated in DEB across different ethnic backgrounds throughout the world and their potential for occurring in the absence of any family history of skin blistering. These mutations were reported to be associated with DDEB-nails only, DDEB-generalized, RDEB and EB pruriginosa (11–13).
Apart from such recurrent mutations, some highly mutable nucleotide positions are also recognized, where different amino acid substitutions result in different clinical presentations. The p. G2073R variation in PT03, PT04 and PT05 is a novel mutation that resulted in a mild DDEB phenotype, though the substitution of the same glycine with aspartic acid (p.G2073D) (14) and valine (p.G2073V) (15) have been previously reported in a patient with RDEB with localized skin lesions and a DDEB-pruriginosa patient, respectively. Similarly, the glycine residue at nucleotide position 1734 that was mutated in PT07 (p.G1734fsX) was previously substituted with aspartic acid (p.G1734Asp) in a patient with DDEB-generalized phenotype (16).
The nonsense variation in PT01 (p.Lys113X) has not been described previously in association with DEB. It affects the von Willebrand factor A (VWFA) domain in the non-collagenous segment (NC-1) of pro-α1 (VII) polypeptide chain (17). Interestingly, this patient’s phenotype is milder in comparison with other patients associated with glycine substitutions in our cohort. Notably, none of our patients with DDEB reported intense pruritus at the time of presentation (or) had a second mutation in the COL7A1 gene. The affected individuals presented with blistering lesions confined to trauma-prone sites, except PT06 and PT07, who showed a more generalized phenotype with blistering predominantly over the lower extremities.
The clinical spectrum of RDEB, on the other hand, is a broad continuum, which can range from a severe blistering condition associated with significant morbidity and functional impairment to a relatively mild disease, clinically indistinguishable from DDEB, as seen in PT18. Although, the clinical features manifest from birth, an accurate classification of the sub-type can often only be made several years after birth, when complications such as syndactyly, mitten deformities, ocular erosions and others develop. Hence, making an early molecular diagnosis and prompt management is of utmost importance to prevent such complications (18).
Consistent with the widely accepted genotype-phenotype correlation concepts, bi-allelic nonsense or splice-site variations resulting in premature termination codons (PTC) were associated with RDEB_GS phenotype (PT08–PT12). Among them, PT09 (W631X) (19) and PT10 (c.3822-2A>G) (20) harboured mutations that were previously reported to be associated with RDEB_GI phenotype in a compound heterozygous state. However, homozygous inheritance in the present cases might explain the severe phenotype observed, consistent with the IFM analysis that revealed completely absent staining for COLVII antibody. Similarly, bi-allelic missense or compound heterozygous missense and nonsense variations were associated with RDEB_GI phenotype (PT14, PT17 and PT18) and showed positive (+ or ++) staining for COLVII at the DEJ.
Noteworthy exceptions to these general rules include PT16, who presented with a RDEB_GI phenotype despite harbouring bi-allelic nonsense variations, and PT13, who presented with extensive muco-cutaneous blistering along with flexion contractures of the fingers, elbows and knees, despite harbouring a previously reported homozygous missense variation (p.G2025S) known to be associated with a mild RDEB phenotype (21). This may be because the patient did not access proper medical care prior to presenting to us at the age of 24 years. Before this, the patient was treated with alternative medicine in his village with no diagnosis of EB.
Similarly, PT15 also presented with a severe clinical phenotype along with partial inter-digital fusion of toes and flexion contractures despite harbouring compound heterozygous missense (p.G2003R) and nonsense (p.R1632X) variations that were previously reported to be associated with DDEB (22) and RDEB_GS (23), respectively. Such phenotypic variations have been attributed to environmental factors and genetic modifiers, such as polymorphisms in matrix metalloproteinase 1 (MMP1) and filaggrin (FLG) genes (24, 25). However, we did not find any polymorphisms in these genes in the present cohort (data not shown). Identifying such sources of disease variation, if any, is crucial to improving biologically valid therapeutic concepts for DEB.
In conclusion, this study describes the phenotype and genotype spectrum of 18 DEB patients from India. Consistent with earlier studies, most patients harboured private mutations, along with a diverse clinical presentation. It is evident that accurate diagnosis of DEB cannot always be predicted precisely based on genetic information and requires IFM studies to provide invaluable information for prognosis and course prediction, especially in infancy before the full phenotype develops. Some mutations and phenotypes are notoriously mild, although they are inherited in a recessive manner. With traditional sequencing, identifying such a second mutation in COL7A1 remains elusive, and hence requires screening of the entire gene, which is often tedious and time consuming. The commercial costs of performing and interpreting WES for EB in India is similar to that reported previously (4), with turnaround times ranging from the shortest of 4–6 weeks to a median of 12–16 weeks, depending on the availability of resources. Targeted panel sequencing for genes implicated in EB and related disorders could prove economic for diagnostic purposes, with rapid turnaround times (5). This preliminary study using NGS-guided diagnosis is clearly encouraging and will serve as a basis for future large-scale molecular studies of EB in the Indian population.
Funding. This study was a part of the PhD dissertation of VKY and was supported by grants obtained by GS from the Department of Science and Technology, New Delhi, India (DST (D-334)) and the Indian Council of Medical Research, New Delhi, India (ICMR (I-817)). VS and SS acknowledge funding from the Council of Scientific and Industrial Research (CSIR), India (grant number BSC0212).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize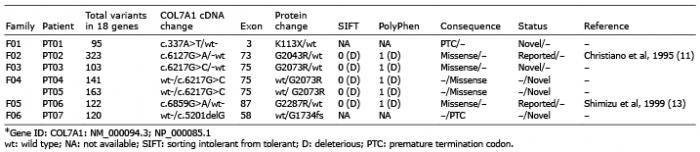 Click to show fullsize
Click to show fullsize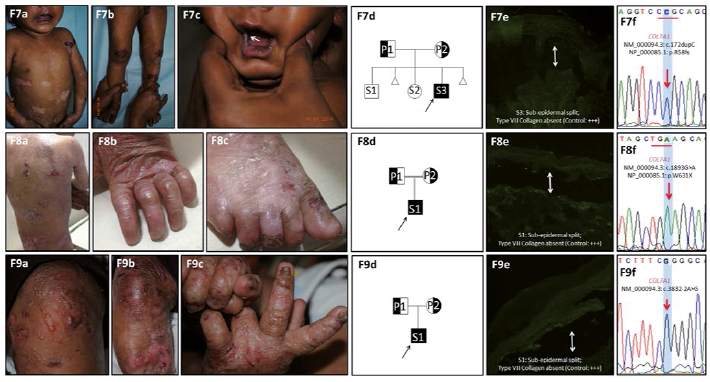 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize