


1Faculty of Medicine, 2Department of Dermatology, 3Institute of Pathology, 4Center of Oncology and Hematology, University Hospital Zurich, Zurich, 5Department of Dermatology, Kantonsspital St Gallen, St Gallen, Switzerland, and 6Department of Dermatology, Kepler University Hospital, Linz, Austria
Rare T- or NK-cell lymphomas with cutaneous manifestation may display a highly aggressive clinical course and major diagnostic/therapeutic challenges. This report describes our experiences with different lymphomas of this rare category and the therapeutic options used. This retrospective, descriptive, monocentric, cross-sectional case study, identified 4 rare aggressive T-/NK-cell lymphomas with manifestation in the skin, which were diagnosed in a tertiary care centre over a period of 4 years. Two patients had an Epstein-Barr virus-associated extranodal NK/T-cell lymphoma and 2 patients had a primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Concomitant extracutaneous involvement was observed in 2 of all 4 patients. Two patients had fulminant disease progression and resistance to chemotherapy. Two patients underwent allogeneic haematopoietic stem cell transplantation, which resulted in one complete remission and one partial remission. This report emphasizes the importance of an early diagnostic work-up and a prompt aggressive therapeutic approach.
Key words: lymphoma; manifestation in the skin; ENKTL; PC-AETCL; allogeneic hematopoietic stem cell transplantation.
Accepted Apr 24, 2018; Epub ahead of print Apr 24, 2018
Acta Derm Venereol 2018; XX: XX–XX.
Corr: Emmanuella Guenova, Department of Dermatology, University Hospital Zurich, Gloriastrasse 31, CH–8091 Zurich, Switzerland. E-mail: emmanuella.guenova@usz.ch
Aggressive T-cell lymphomas with primary manifestation in the skin are rare and often harbour major diagnostic/therapeutic challenges. In this retrospective descriptive monocentric cross-sectional case study over a period of 4 years, we identified a total of 4 patients suffering from aggressive T-cell lymphomas with primary manifestation in the skin. This corresponded to < 1% of all newly diagnosed cutaneous lymphoma cases. We observed an aggressive disease course in all our patients and disease progression and resistance to chemotherapy in two of them. Our report emphasizes the importance of an early extensive diagnostic work-up and a prompt aggressive therapeutic approach.
Mature T- and NK-cell malignancies are rare. However, in the skin, approximately 75% of all cutaneous lymphoma are of T-cell (approximately 65%) and NK-cell (approximately 10%) origin (1–5). Most of the T-cell lymphomas manifesting in the skin are primary cutaneous, i.e. originating from cells with skin-homing capacity that either permanently resides in the skin or recirculates through the blood (6, 7). Skin manifestation occurs in rare cases as the first clinical appearance of peripheral T-cell lymphomas (8, 9).
The 2008 WHO classification, revised in 2016, recognizes extranodal NK/T-cell lymphoma (ENKTL) as one of the prototypes of virally associated Epstein-Barr virus (EBV)-positive T-cell or NK-cell lymphoma (9–12). ENKTL, nasal type affects adult individuals and is more frequent in Asia, Mexico and South America, where it accounts for up to 6% of all cases of non-Hodgkin’s lymphomas (13). Males are affected 2–3 times more often than females (14). ENKTL nasal type manifests mostly in the nasal/paranasal area and can further involve the skin, gastrointestinal tract and, in rare cases, the bone marrow (15). An extranasal manifestation of ENKTL is less common, resulting in the suggestion of some authors that, in most cases of extranasal ENKTL, an occult nasal involvement was missed on initial evaluation/staging (9, 16–18). The prototypic immunophenotype of ENKTL is CD2+CD56+. Surface CD3 and other common T-cell antigens (CD4, CD8, CD5) are usually negative with positivity for cytoplasmic CD3ε. Some tumours express α/β-T-cell receptor (α/β-TCR). γ/δ TCR expression has rarely been reported and molecular analysis only rarely detects a monoclonal rearrangement of the TCR gene (19). Overall patient survival and clinical features seem to be similar in T- and NK-cell phenotype ENKTL (20). Characteristically, EBV can be detected in almost all cases of ENKTL (21).
The prognosis of ENKTL depends on the disease stage. Disease manifestation outside the site of initial tumour manifestation is associated with poor prognosis (15). Elevated levels of lactate dehydrogenase and systemic symptoms of fever, malaise and weight loss are additional, independent poor prognostic factors (22).
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (PC-AETCL) has been recognized as a provisional entity in the 2016 revision of the WHO classification of lymphoid neoplasms (9, 12). CD8+ cytotoxic T lymphocytes with pronounced epidermotropism and an aggressive clinical course are hallmarks of this lymphoma entity. The tumour affects adult individuals, but a case of primary cutaneous CD8+ PC-AETCL in a child has been reported (23). The prototypic immunophenotype of this lymphoma entity is CD3+/CD4–/CD8+ with monoclonal rearrangement of the TCR gene. Tumour cells express cytotoxic molecules (TIA-1+, granzyme B+) and the α/β-TCR. NK cell markers (CD56) and γ/δ TCR are not expressed, and EBV is not detectable, in neoplastic cells. In some cases, expression of pan-T-cell-markers may be lost (24). The estimated 5-year survival of patients with PC-AETCL is 0%.
Other rare types of aggressive NK/T-cell lymphomas that can primarily manifest in the skin include angioimmunoblastic T-cell lymphoma, primary cutaneous γ/δ T-cell lymphoma and severe hydroa vacciniforme-like T-cell lymphoma (a sub-entity in the group of hydroa vacciniforme-like lymphoproliferative disease) (25–27).
The diagnostic and therapeutic handling of these rare aggressive cutaneous T- and NK-cell lymphomas represents a major hurdle in clinical practice. The aim of this report is to describe our experiences with different lymphoma cases of this category, emphasizing their very particular clinical features and/or therapeutic approaches.
For this retrospective, descriptive, monocentric, cross-sectional case study, a comprehensive search of the patients’ data repository of a tertiary care centre covering a population of approximately 1.6 million people (University Hospital Zurich: approximately 1.5 million; Kantonsspital St Gallen: approximately 0.1 million) was performed for the period between 1 January 2013 and 1 January 2017. All identified patients with aggressive rare T-cell lymphomas with primary manifestation in the skin (definition according to the International Classification of Diseases, Tenth Revision code C84.4) were included in the study. We identified a total of 4 individuals with aggressive rare T-cell lymphomas with primary manifestation in the skin.
Between 1 January 2013 and 1 January 2017 a total of 53 cutaneous T-cell lymphomas were diagnosed at our tertiary cutaneous lymphoma centre, which covers a population of approximately 1.6 million people (incidence: 0.83 per 100,000 person-years). Four of these were aggressive rare T-cell lymphomas with primary manifestation in the skin, thus accounting for < 1% of all newly diagnosed cutaneous T-cell lymphomas. The following 4 aggressive rare T-cell lymphomas with primary manifestation in the skin were identified (Table I): (i) EBV-associated ENKTL nasal type with γ/δ TCR expression; (ii) EBV-associated extranasal ENKTL; (iii) PC-AETCL with an aberrant phenotype; and (iv) PC-AETCL. The mean age of the patients was 67.75 years (ranging from 51 to 87 years). Three out of 4 patients were female, 1 was male. The mean time from the first clinical symptoms to final diagnosis was 5.75 months (range 2–12 months). All patients had an initial skin manifestation, with a concomitant extracutaneous involvement of the spine, bone marrow and/or lymph nodes in 2 out of 4 cases.
In 2 of the patients, fulminant disease progression and resistance to chemotherapy led to the patients’ death 64 and 93 days after diagnosis, respectively. In 2 of the patients, allogeneic haematopoietic stem cell transplantation (HSCT) resulted in complete/partial remission for 9 months and 18 months followed by a relapse. Apart from mild mixed acute and chronic graft-versus-host-disease (GVHD) in one of the patients, no serious transplantation-related complications have occurred.
Table I. Patient characteristics and disease courses
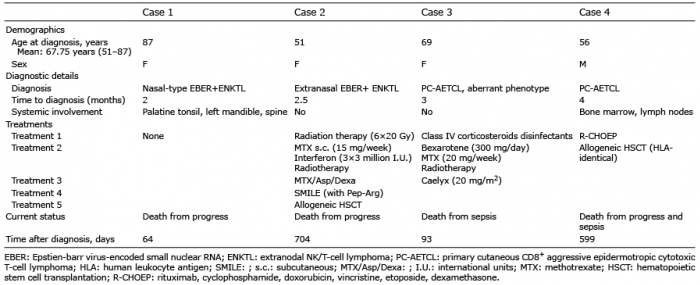
An 87-year-old woman presented with necrotizing gingivitis of unknown origin. She reported an approximately 2-month history of painful oral aphtous stomatitis and gingival bleeding. Initial treatment with disinfecting rinsing agents (for aphtous stomatitis) remained ineffective. Clinical examination (Fig. 1a) revealed a solitary greyish, infiltrated and ulcerated plaque on the upper palate and multiple enoral small aphtous ulcerations. An enoral biopsy was performed (Fig. 1b), which showed a CD3+/CD2+, cytotoxic (perforin+/granzyme B+/TIA1–/TdT–), highly proliferative infiltrate. The cells exhibited partial (30%) co-expression of CD30 and CD56, but were completely negative for CD4, CD8, CD5 and CD7. TCRγ/δ gamma (but not TCR-β-F1) and EBV-encoded small nuclear RNA (EBER) was found on/in the tumoural cells, thus allowing us to diagnose an EBV-associated ENKTL nasal type with γ/δ TCR expression. Positron emission tomography–computed tomography (PET-CT) showed multiple metabolically active focuses and bone lesions (spine).
Because of the aberrant CD30 expression, we aimed for systemic treatment with brentuximab-vedotin, an antibody-drug conjugate of an anti-CD30 monoclonal antibody and the proapoptotic anti-tubulin agent monomethyl auristatin E (28–30). However, the patient declined any lymphoma-specific intervention, and died from disease progression (Fig. 1c) and sepsis 64 day after diagnosis.
Fig. 1. Case 1. (A) Initial clinical presentation. (B) Immunohistopathological features (HE: haematoxylin and eosin) and expression of immunophenotypical markers. (C) Clinical presentation after 3 weeks.
A 51-year-old woman presented with a corticosteroid-resistant nodule on her right lower leg. The patient was otherwise asymptomatic and related the nodule to a mosquito bite received a few weeks earlier. Clinical examination (Fig. 2a) showed a solitary erythematous subcutaneous nodule on the right calf, which was firm and painful on palpation. There was no additional cutaneous involvement and no lymphadenopathy.
Skin biopsy (Fig. 2b) revealed a small-to-medium-sized pleomorphic, highly proliferative CD3+/CD2+ T-cell population with CD8 expression in approximately 30% of all T cells. Skin flow cytometry (Fig. 2d) demonstrated a clearly defined CD16+/CD56+ NK-cell population. Molecular analysis did not detect any clonal rearrangement of the TCR, immunohistochemistry was negative for TCR γ/δ or β-F1 (TCR α/β) and EBV in situ hybridization was highly positive. PET-CT excluded nasal involvement and systemic dissemination, confirming our diagnosis of an extranasal EBV+ ENKTL.
Following initially successful radiation therapy (6×20 Gy), a local relapse occurred (Fig. 2a). Our subsequent therapeutic approaches with combined methotrexate/interferon/percutaneous radiotherapy and methotrexate/asparaginase/dexamethasone remained ineffective. Following 2 cycles of SMILE chemotherapy (dexamethasone, methotrexate, ifosfamide, Peg-asparaginase, and etoposide) and an allogeneic HSCT with reduced intensity conditioning, the patient achieved a partial remission, accompanied by an acute and, subsequently, chronic cutaneous GVHD (superficial sclerosis, lichenoid oral involvement). The patient remained relapse-free for 18 months (Fig. 2c) before a massive pericardial effusion with tumour cells occurred and she died from progressive disease.
Fig. 2. Case 2. (A) Initial clinical presentation. (B) Immunohistopathological features (HE: haematoxylin and eosin) and expression of immunophenotypical markers. (C) Clinical presentation after 2.5 years (left), lichenoid features of cutaneous graft-versus-host disease.
A 69-year-old woman presented with newly developed generalized ulcerations and pruritus. The patient reported a 3-month history of weight loss and night sweats. Multiple sharply demarcated targetoid erythematous patches and plaques were found on the trunk and extremities, most of them centrally ulcerated, and painful upon palpation (Fig. 3a). Mucosal sites were not involved.
Skin biopsy (Fig. 3b) revealed a prominent lymphocytic infiltrate exhibiting an aberrant cytotoxic phenotype. The tumour cells were negative for CD3, CD4, CD30, CD7 and CD56, and EBV (EBER1) with low CD8, but high expression of the cytotoxic molecules perforin and granzyme B. TCR genotyping revealed 2 clonal fragments (201/202bp and 204/205bp). Chromosomal translocation analysis was unremarkable. A PC-AETCL was diagnosed with an aberrant immune phenotype and partial loss of CD8 expression. Examination of the peripheral blood and the bone marrow showed no abnormalities, and a total body CT further excluded extracutaneous organ involvement (Fig. 3d). Skin-directed treatment with class IV topical corticosteroids, polidocanol and disinfectant bathes remained ineffective. Administration of bexarotene (300 mg/d) and methotrexate (20 mg/week) combined with local radiotherapy resulted in partial healing of the lesions. Two months later, the patient developed generalized painful, centrally necrotic plaques (Fig. 3c) with high metabolic activity in PET-CT. Following 1 cycle of chemotherapy with pegylated liposomal doxorubicin (20 mg/m2), her condition deteriorated and she developed new ulcerating lesions, extensive oral candidiasis, fever, leukocytopaenia and hyperchromic macrocytic anaemia. The patient died from a skin infection with S. aureus and subsequent sepsis 93 days after the initial diagnosis of PC-AETCL.
Fig. 3. Case 3. (A) Initial clinical presentation. (B) Immunohistopathological features (HE: haematoxylin and eosin) and expression of immunophenotypical markers. (C) Clinical presentation after 3 months. (D) Positron emission tomography–computed tomography (PET-CT) at initial staging and 3.5 months after diagnosis.
A 56-year-old man presented with disseminated ulcerating skin nodules, oral necrotic ulcerations and axillary and cervical lymphadenopathy. The oral ulcerations had appeared 4 months earlier and had been treated unsuccessfully with disinfecting solution. The lesions on the trunk and extremities had developed approximately 1 month later (Fig. 4a). In addition, the patient had a 2-week history of decreased appetite, weight loss and night sweats. The detailed medical history revealed chronic lymphocytic leukaemia followed up with watchful waiting during the last 6 years. Clinical examination (Fig. 4a) revealed disseminated, painful, centrally ulcerating nodular lesions (up to 5 cm in size), one large oral necrotic plaque and generalized lymphadenopathy.
A skin biopsy (Fig. 4b) revealed a CD3+/CD8+/CD4–/TCRβ–F1+ proliferative infiltrate of small atypical lymphocytes accumulating in the perivascular and periadnexal areas. Molecular analysis demonstrated a clonal rearrangement of the TCR. PET-CT (Fig. 4d) revealed multiple metabolically active cutaneous and subcutaneous lesions and generalized lymphadenopathy. Histology confirmed further T-cell lymphoma involvement in the lymph nodes and in the bone marrow. The patient was diagnosed with a PC-AETCL as a composite lymphoma with B-cell chronic lymphocytic leukemia (CLL) and initiated treatment with R-CHOEP (rituximab, cyclophosphamide, doxorubicin, vincristine, etoposide and dexamethasone). After 6 cycles, the patient achieved complete remission, which allowed for a reduced conditioning regimen (fludarabine 30 mg/m2, busulfan 4×1 mg/kg and ATG 10 mg/kg), and an allogeneic HSCT of a human leukocyte antigen (HLA)-identical donor (the patient’s brother). The first 9 months of follow-up were uneventful (Fig. 4c, d). Afterwards, the CLL relapsed and the patient developed a spontaneous bacterial peritonitis with a septic shock and died.
Fig. 4. Case 4. (A) Initial clinical presentation. (B) Immunohistopathological features (HE: haematoxylin and eosin) and expression of immunophenotypical markers. (C) Clinical presentation after 4 months. (D) Positron emission tomography–computed tomography (PET-CT) at initial staging and after 6 months following haematopoietic stem cell transplantation.
This monocentric cross-sectional case study aimed to summarize our experience with aggressive rare T-cell lymphomas with skin manifestation. In 4 consecutive years, we observed 4 cases: 2 with ENKTL and 2 with PC-AETCL. All cases harboured major diagnostic and therapeutic challenges.
The particular oral manifestation of the ENKTL (case number 1) and the PC-AETCL (case number 4) led to a significant diagnostic and therapeutic delay. In contrast, the cutaneous ENKTL nodule (case number 2) and the fulminant skin ulcerations of the PC-AETCL (case number 3) were clinically much more suggestive of lymphoma and thus immediately biopsied (31). This emphasizes the importance of a rapid histological examination on encountering treatment-refractory oral lesions.
Treatment modalities for ENKTL include radiotherapy, chemotherapy, or a combination of both (32). L-asparaginase has shown good in vitro anti-NK-cell lymphoma activity, and the introduction of chemotherapeutic regiments including L-asparaginase has improved the outcome or ENKTL (32). Allogeneic HSCT is reserved for patients with relapsed/refractory disease; in a cohort of 18 patients, allogeneic HSCT resulted in a 5-year-progression-free survival of 57% (33). Our patient with ENKTL (case 2) achieved and maintained remission for approximately 18 months.
In terms of therapy with the anti-CD30 brentuximab vedotin, the significance of CD30 expression in ENKTL is still under debate. While Feng et al. found that CD30 expression (observed in 47.3% of a cohort of 91 patients) did not correlate with clinicopathological or prognostic features (34), Wang et al. reported an association with shorter overall and progression-free survival (35). The use of the anti-CD30 brentuximab vedotin has resulted in complete remission in 2 cases reported so far (28, 30).
PC-AETCLs constitute a rare, poorly characterized subgroup of cutaneous lymphoma and are, according to the 2016 WHO classification, still considered as a provisional entity (9). A recent analysis of data from 34 patients with PC-AETCL confirms poor prognosis, with a 5-year survival rate of 32% and a median survival of only 12 months (36). Autologous/allogeneic HSCT has shown promising results in several recently published cases (36, 37). One of our 2 PC-AETCL patients (case number 3) did not respond well to treatment with bexarotene, methotrexate, local radiotherapy and pegylated liposomal doxorubicin and died of progressive disease and sepsis. Our second patient with PC-AETCL (case number 4) achieved a remission of at least 9 months after R-CHOEP and allogeneic HSCT.
In conclusion, our experience with rare ENKTL and PC-AETCL emphasizes the importance of an early extensive diagnostic work-up and prompt aggressive therapeutic approach including allogeneic HSCT.
This study was supported by the Helmut Horten Stiftung (to E.G.), the Promedica Stiftung (1406/M and 1412/M to E.G.), the Krebsliga Schweiz (KFS-4243-08-2017 to E.G.), the Forschungskredit of the University of Zurich (FK-15-040 to W.H. and FK-17-023 to Y.T.C.), the Jubiläumsstiftung von SwissLife (to E. G.) and the Hochspezialisierte Medizin Schwerpunkt Immunologie (HSM-2 Immunologie to W.H.) Schweiz. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize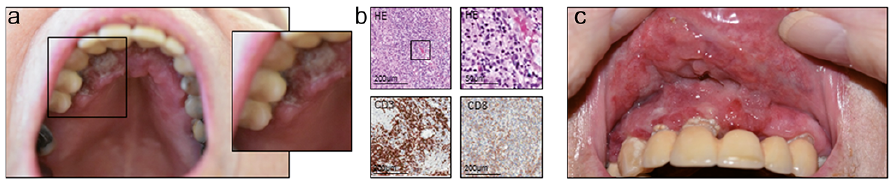 Click to show fullsize
Click to show fullsize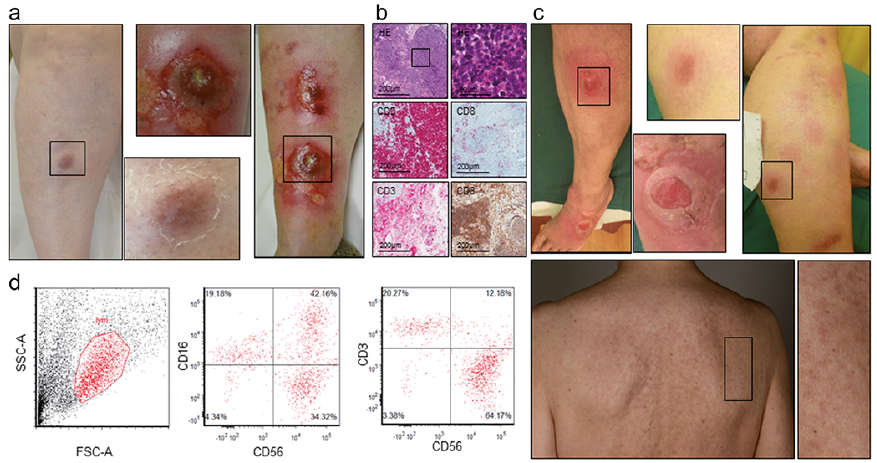 Click to show fullsize
Click to show fullsize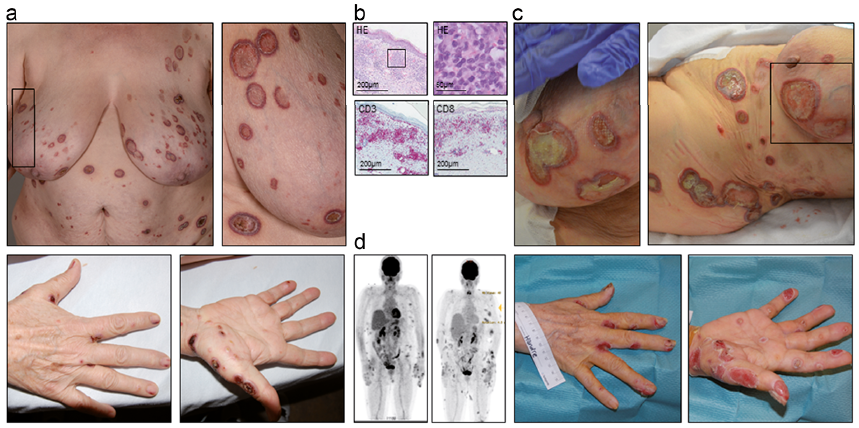 Click to show fullsize
Click to show fullsize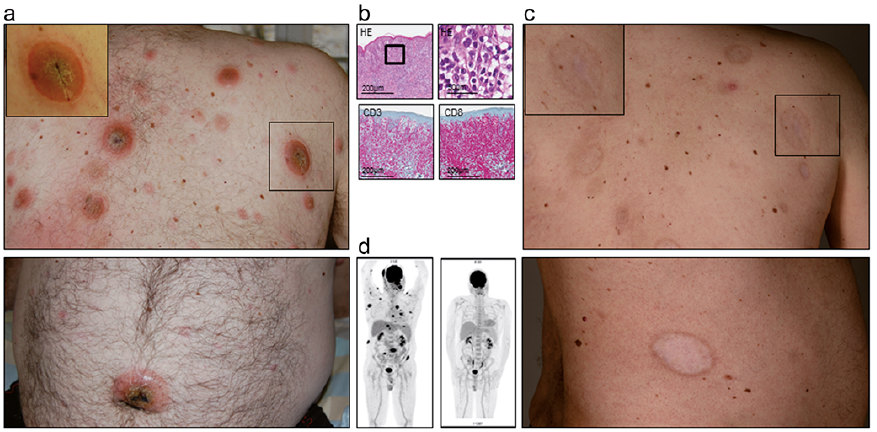 Click to show fullsize
Click to show fullsize