


1King’s College London, St John’s Institute of Dermatology, London, UK, 2Université Grenoble Alpes, SyMMES, & CEA, INAC, LAN, Grenoble, 3Pierre Fabre Dermo-Cosmétique, Clinical Skin Research Center, and 4Dermatology Department, Larrey University Hospital, Toulouse, France
The cyclobutane pyrimidine dimer (CPD) is a potentially mutagenic DNA photolesion that is the basis of most skin cancers. There are no data on DNA protection by sunscreens under typical conditions of use. The study aim was to determine such protection, in phototypes I/II, with representative sunscreen-user application. A very high SPF formulation was applied at 0.75, 1.3 and 2.0 mg/cm2. Unprotected control skin was exposed to 4 standard erythema doses (SED) of solar simulated UVR, and sunscreen-treated sites to 30 SED. Holiday behaviour was also simulated by UVR exposure for 5 consecutive days. Control skin received 1 SED daily, and sunscreen-treated sites received 15 (all 3 application thicknesses) or 30 (2.0 mg/cm2) SED daily. CPD were assessed by quantitative HPLC-tandem mass spectrometry (HPLC-MS/MS) and semi-quantitative immunostaining. In comparison with unprotected control sites, sunscreen significantly (p ≤ 0.001–0.05) reduced DNA damage at 1.3 and 2.0 mg/cm2 in all cases. However, reduction with typical sunscreen use (0.75 mg/cm2) was non-significant, with the exception of HPLC-MS/MS data for the 5-day study (p < 0.001). Overall, these results support sunscreen use as a strategy to reduce skin cancer, and demonstrate that public health messages must stress better sunscreen application to get maximal benefit.
Key words: sunscreen; photoprotection; DNA protection; cyclobutane pyrimidine dimers.
Accepted Jun 14, 2018; Epub ahead of print Jun 25, 2018
Acta Derm Venereol
Corr: Antony R Young, King’s College London, 9th Floor, Tower Wing, Guy’s Hospital, London SE1 9RT, UK. E-mail: antony.young@kcl.ac.uk
Skin cancer is an increasing public health burden in many countries. Most skin cancers are caused by DNA damage from ultraviolet radiation in sunlight. This study shows that a very high sun protection factor sunscreen can inhibit DNA damage in the skin caused by high doses of artificial sunlight, even when the sunscreen is used less than optimally. The data suggest that sunscreen use is likely to reduce skin cancer and that there should be more emphasis in communicating how to best use sunscreens in public health campaigns.
Solar ultraviolet radiation (UVR) causes skin cancers derived from epidermal melanocytes (melanoma) and keratinocytes (basal cell carcinoma (BCC) and squamous cell carcinoma (SCC)). Skin cancer incidence is increasing in many countries with predominantly fair-skinned populations (1, 2). Epidemiology has shown a relationship between sunburn (erythema) and malignant melanoma (MM), especially with childhood exposure (3). There is also evidence for such a relationship for BCC. The epidemiology for SCC supports a role for chronic low dose (sub-erythemal) solar UVR exposure (4).
Keratinocyte cancers (KC) are initiated by UVR-induced DNA damage, in particular the cyclobutane pyrimidine dimer (CPD) that results in characteristic C to T transition mutations in key regulatory genes such as p53 (5). There is evidence for a role for such mutations in MM (6), though its molecular pathogenesis is more complex.
Sunscreens increase the dose required to induce ery-thema for which their index of efficacy is the sun protection factor (SPF) that is determined by exposing the skin to solar simulated radiation (SSR) with the sunscreen applied at 2 mg/cm2. However, sunscreens may not fully inhibit sunburn (7, 8). This is because people typically overestimate protection indicated by the label, by using much less sunscreen than 2 mg/cm2 with a commensurate reduction of actual SPF (9).
Sunscreen use is widely advocated as a means of reducing skin cancer risk and this has been supported by a randomised trial of sunscreen use (SPF 16) which showed protective effects for actinic keratoses (AK) (10), and also for SCC (11) in a high-risk population in sub-tropical Australia. To date, there is no convincing evidence that sunscreen use has any significant impact on BCC. Meta-analyses of several case-control studies have shown no effect of sunscreen use on melanoma (12). However, more recently, a reduction of melanoma in the same Australian study population as described above (13) and also a large prospective population-based study reported that use of sunscreens with SPF ≥ 15 vs SPF < 15 reduced the risk of melanoma (hazard ratio 0.67 (95% confidence interval (CI) 0.53–0.83) in Norway (14). Long-term sunscreen use has also been shown to inhibit photoageing (15).
Prospective trials for skin cancer prevention by sunscreens are complex and the International Agency for Research on Cancer (IARC) identified the need for sunscreen studies on biomarkers, such as DNA damage, that are predictive of cancer risk (16). A few studies have assessed the ability of sunscreens at 2 mg/cm2 to inhibit DNA photodamage in human skin in vivo but some have design flaws and other limitations (recently reviewed by Olsen et al. (17)), such as the use of non-solar UVB (280–320 nm) sources, sampling too late after UVR exposure (e.g. 24–48 h and longer), when many/most lesions will have been repaired (18, 19), and using the same low UVR challenge dose for sunscreen-protected and unprotected skin that is a very undemanding test for a sunscreen.
However, no study has assessed the effect of lowering sunscreen application thickness below 2 mg/cm2 on DNA protection, or on photoprotection in the basal layer that contains keratinocyte stem cells and melanocytes. We report on studies of which the primary aim was to assess the ability of a very high SPF sunscreen to inhibit overall epidermal CPD in sun sensitive skin types I/II after (i) a single SSR exposure and (ii) repeated SSR exposures to simulate a short holiday at tropical latitude. The secondary aim was to assess CPD in basal cell keratinocytes and melanocytes. Good ethical practice required that SSR doses on unprotected sites be sub-erythemal or borderline erythemal.
This study, approved by the Cambridge South, UK, Ethics Committee (ref: 13/EE/0268), was conducted in accordance with the Declaration of Helsinki, at St John’s Institute of Dermatology, London, UK from December 2013 to April 2014.
People use sunscreens to extend their intentional solar exposure time (20) so higher, but realistic, SSR doses were used on protected sites to ensure a robust test of the sunscreen in question. DNA photodamage was assessed, in the same skin samples, by quantitative mass spectrometry and semi-quantitative immunostaining that enables damage location within the epidermis. The thymine dimer (TT) was chosen as the primary biomarker because it is the most frequent CPD (21, 22), but other di-pyrimidine lesions were also quantified in the acute study. Biopsies were taken immediately after irradiation (last irradiation in repeated exposure study) to minimise any confounding impact of DNA repair.
Sixteen healthy young phototype I and II volunteers were recruited by internal advertisement and gave written informed consent before participation. There were two groups, each with 8 persons (3 women, 5 men with one skin type I, and 7 skin types II). One group received a single UVR exposure (hereafter called the acute exposure group). The other received exposures on 5 consecutive days (hereafter called the repeated exposure group). The mean ± standard deviation (SD) ages of the acute and repeat exposure groups were 23 ± 3.7 and 25 ± 3.5 years, respectively.
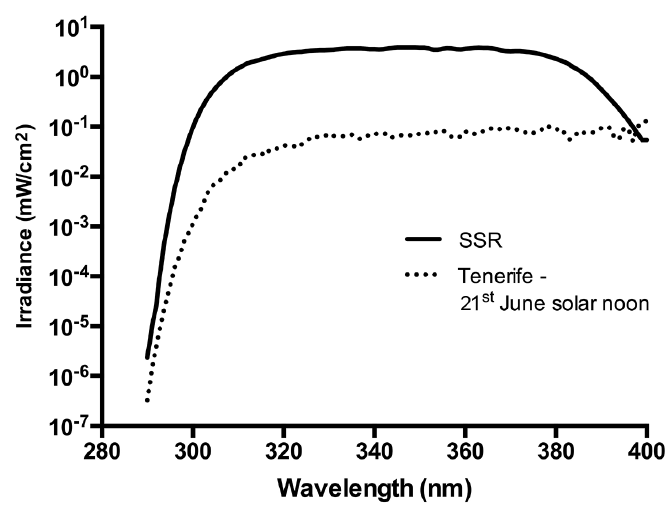
SSR was obtained from a Solar® Light 16S-001 v4.0 (Solar® Light, Glenside, Pennsylvania) and delivered by a liquid light guide (7 mm exit diameter). The irradiance was spectroradiometrically determined (23) and the emission spectrum (Fig. 1) was compliant with the International Organization for Standardisation (ISO) Standard 24444 and Cosmetics Europe 2006. Irradiance was measured routinely with a Solar® Light PMA 2100 radiometer (Solar® Light, Glenside, Pennsylvania) after calibration by spectroradiometry. Exposure was expressed as standard erythema doses (SED). One SED was typically delivered in about 6 s. For comparison, Fig. 1 also shows a simulated noon mid-summer solar spectrum from Tenerife (28.3° N, 16.6° W) (24). The SSR spectrum contains relatively less UVA1 (340–400 nm) than natural sunlight, but the overall UVA (320–400nm) erythemally effective energies (EEE) of each spectrum were similar; SSR = 13.2% and Tenerife = 15.2%.
Fig. 1. Emission spectrum of solar simulated radiation (SSR) source compared with simulated noon solar summer solstice spectrum in Tenerife (22). The UVB/UVA components of the spectra are 10.7/89.3% (SSR) and 6.2/93.8% (Tenerife), corresponding respectively to 86.8/13.2% and 84.8/15.2% of the erythemally effective energy.
Sunscreen and application. The sunscreen (SPF 50+) was provided by Laboratoires Pierre Fabre (Toulouse, France). It complied with all relevant EU regulations with no evidence of phototoxicity. The filters were bis-ethylhexyloxyphenol methoxyphenyl triazine (Tinosorb S®), diethylhexyl butamido triazone (Uvasorb®HEB), butyl methoxydibenzoyl methane (Parsol®1789) and methylene bis-benzotriazolyl tetramethylbutylphenol (Tinosorb M®). The product also contained the antioxidant tocopheryl glucoside (pre-tocopheryl®) (25). The mean ± SD SPF (2 mg/cm2) was 64.0 ± 15.8 and the in vitro UVA protection factor (UVA-PF) (ISO 24443 Standard) was 25.3 with a critical wavelength of 379 nm. The mean ± SD SPFs at 0.75 and 1.3 mg/cm2 were 20.9 ± 3.3 and 42.5 ± 8.4, respectively (Pierre Fabre Dermo-cosmetique internal files).
The study site was the sun-protected upper buttock in which 1 minimal erythema dose (MED) of SSR is ~3 SED in types II (23). The sunscreen was applied to three 5 × 7 cm2 zones of skin to achieve target application thicknesses of 2 mg/cm2, 1.3 mg/cm2 and 0.75 mg/cm2. Each volunteer had a personally pre-assigned tube of sunscreen. The product was decanted onto a weighing boat per test site/per person and the boat was re-weighed after application. The sunscreen was “spotted” evenly over the application area and then lightly spread transversely and then perpendicularly. This was done with a finger cot pre-applied with a small amount of sunscreen to prevent loss by absorption by the cot and a new finger cot was used for each sunscreen thickness. The mean ± SD amounts (mg) applied (over 35 cm2) in the acute exposure study were 70.38 ± 0.74, 45.01 ± 0.54 and 26.03 ± 0.72 corresponding to 2.01, 1.29 and 0.74 mg/cm2, respectively. Comparable daily quantities were achieved in the repeated exposure study. Summaries of the experimental set-up are shown in Fig. 2. In the acute exposure group, the sunscreen treated sites were exposed to 30 SED (~3 h tropical exposure). A positive control site, not treated with sunscreen, was exposed to 4 SED and a completely untreated site served as a negative control. In the repeated SSR exposure group, treatments were given for 5 consecutive days. The daily dose was 15 SED with the sunscreen at 1.3 mg/cm2, 0.75 mg/cm2 and 2 mg/cm2. An additional site with the sunscreen at 2 mg/cm2 was exposed to 30 SED/daily. One positive control site was exposed to 1 SED daily with no sunscreen. Irradiations were within 15 min of sunscreen application in both groups. The same procedure, dosing and application density was used for skin phototypes I and II. Sites were not randomised but all assessments of DNA damage were undertaken blinded.
Fig. 2. Summary of experimental set-up. (a) Acute exposure group and (b) repeated exposure group. Sunscreen application thicknesses given as mg/cm2 and solar simulated radiation exposures as standard erythema doses (SED). The biopsies were split for HPLC-tandem mass spectrometry (HPLC-MS/MS) and immunostaining assays with the exception of the acute exposure group exposed to 30 SED with the sunscreen at 2mg/cm2 for which there were duplicate sites; one for HPLC/MS-MS and one for immunostaining.
Punch biopsies (4 mm) were taken under local anaesthetic from the centre of each treated site. This was within 15 min of the single and 5th irradiations in the acute and repeated exposure groups respectively.
The biopsies were trimmed to remove most of the dermis and divided into two halves. One half was analysed by immunostaining and the other by HPLC-tandem mass spectrometry (HPLC-MS/MS). There was one exception to this with the acute study, for which there was a full biopsy for each type of assay with the sunscreen application at 2 mg/cm2. Tissue for immunostaining was paraffin-embedded and cut into 4 µm sections. Tissue for HPLC was immediately snap frozen in liquid nitrogen and stored at –80°C until transport on dry ice (temperature logged) to Grenoble, France and stored at –80°C until analysis.
The techniques have been previously described (26). Results were expressed as the number of bipyrimidine photoproducts/106 normal bases. The main analysis was for TT because they are the most frequent CPD. However, in the acute group, we also analysed TC and CT CPD, as well as TT and TC pyrimidine (6-4) pyrimidone photoproducts (6-4PP) and their Dewar photoisomers.
Sections were deparaffinised with xylene and microwaved for 12 min in citrate buffer (pH6) (Sigma Aldrich, St. Louis, USA). Slides were then submerged in cold running tap water for 3 min, further washed twice in phosphate-buffered saline (PBS) and incubated in 0.6% H202 and 0.1% triton X-100 in PBS for 10 min. Slides were washed again in PBS, incubated for 45 min in blocking buffer (10% goat serum, 0.1% bovine serum albumin and 0.1% tween-20 in PBS) and further incubated with 2 monoclonal antibodies overnight at 4°C. The 2 primary antibodies were Anti-CPD (Clone TDM-2) (Cosmobio, Tokyo, Japan) at 1:1000 and Anti-TRP1 (Abcam, Cambridge, UK) at 1:100 to identify melanocytes. Slides were washed twice in PBS and incubated with 2 secondary antibodies: Alexa Fluor goat anti-mouse 488 (Invitrogen, Paisley, UK) and Alexa Fluor goat anti-rabbit 555 (Invitrogen, Paisley, UK), both at 1:200 for 30 min. Slides were then washed 3 times (twice in PBS and once in distilled water) counterstained with prolong gold anti-fade with DAPI, cover-slipped and left at room temperature in the dark for 2 h prior to image capture.
Image capture was carried out using a Zeiss Axio-Observer Z1 Microscope (Carl Zeiss, Cambridge, UK) and AxioVision Rel. 4.8 software (Carl Zeiss). All fluorescent channel exposures were measured at the beginning of each batch of pictures taken and remained constant throughout each experiment. All samples from a given volunteer were stained in the same batch, to minimize the effects of variation between stain runs, and all pictures from a given batch were captured at the same time. Three sections of each biopsy were stained and 3 photographs of each section were taken. We captured ~80 nuclei per photograph (termed whole epidermis) including ~20 basal keratinocytes and ~3 melanocytes, that were also assessed separately.
Analysis was carried out using AxioVision Rel. 4.8 software (Carl Zeiss, Cambridge, UK). Analysis parameters were established at the beginning of each sample batch to set the upper and lower limits of signal, which eliminated as much background staining as possible. For each individual picture, the regions included in analysis were checked and any dermal artefacts/cells were deleted. In the case of melanocyte/basal layer analyses all cells apart from those being analysed were deleted. The software gave a value for the mean intensity (grey scale) of each picture; i.e. TT/picture. Values for the positive controls of each person (SSR without sunscreen) were set at 100% and all other values were expressed as a percentage of this value because it was not possible to stain all biopsies in a single batch.
The data were analyzed parametrically with STATA v12 (StataCorp, College Station, Tx, US). The endpoints for the statistical assessment were %TT CPD relative to the no-sunscreen positive control (set at 100%) for the immunostaining markers, and number of TT CPD/106 normal bases in the HPLC-MS/MS assay. The negative controls (0 SED) sometimes gave a low level of background noise. A paired t-test was used to demonstrate the effect of SSR by comparing positive and negative controls. A one-sample t-test was used when the reference values were either 0% (i.e. no background noise) or 100%. Some sites treated with sunscreen and SSR had zero TT CPD values (DNA damage below the detection limit), which were included in the calculations because they show a high level of protection. The difference across experimental treatments was analysed using a repeated one-way ANOVA for 3 or more conditions. When ANOVA results suggested differences in mean values (p < 0.05) post hoc tests were performed to determine where these differences lay. The p values were adjusted for multiple comparisons using a Bonferroni correction, with significance set at p ≤ 0.05. The effect of sunscreen application thickness on the endpoints was assessed with a generalised estimating equation (GEE) approach to linear regression.
Table I shows the results for all bipyrimidine photoproducts assessed by HPLC-MS/MS. There were 31.1 ± 14.8 TT CPD/106 bases with 4 SED compared with 6.0 ± 6.4 with 30 SED and sunscreen at 2.0 mg/cm2. Thus, the sunscreen treated sites, with a 7.5 fold higher SSR dose, had ~20% of the DNA damage of the positive control. Fig. 3 shows representative immunostaining, with more CPD after 4 SED without sunscreen than 30 SED with sunscreen at 0.75 and 2.00 mg/cm2. All TT CPD data are shown in Fig. 4. The HPLC technique could not detect TT CPD in 3/8 and 5/8 test sites with sunscreen at 2.0 and 1.3 mg/cm2 respectively (zero values included in the analyses), as was the case with immunostaining in some basal and melanocyte assessments (≤ 3/8). There were significantly (p < 0.001) more TT CPD at 4 SED than 0 SED for all endpoints. ANOVA of all SSR treated sites/assays showed p < 0.001, except for melanocytes with p = 0.045. Table II shows the results of the Bonferroni-adjusted post hoc tests. There was significantly more damage for all endpoints with 4 SED without sunscreen than with 30 SED with sunscreen at 1.3 and 2.0 mg/cm2. The damage was also lower with the sunscreen at 0.75 mg/cm2 but the difference was not significant for any endpoint. ANOVA of the sunscreen treated sites/assays alone gave p-values from < 0.001 to 0.014 for all endpoints, except for melanocytes (p = 0.254). The Bonferroni adjusted post hoc test p-values for the comparisons between sunscreen treated sites show significant differences between 0.75 mg/cm2 and the other two application thicknesses (p < 0.001–0.035), but no differences between 1.3 and 2.0 mg/cm2 (p = 0.145–0.999). The GEE analysis (Table II) showed a significant linear trend for TT CPD protection with increasing sunscreen application thickness except for melanocytes.
Table I. Mean levels of dipyrimidine photoproducts/million normal base pairs in the acute exposure group
Fig. 3. Representative immunostaining of epidermal thymine dimers (TT). DAPI stained blue identifies nuclei and nuclear TT are stained red. Acute group after (a) 4 standard erythema doses (SED) with no sunscreen, (b) 30 SED with sunscreen at 0.75 mg/cm2, (c) 30 SED with sunscreen at 2.0 mg/cm2, which shows a very high level of protection.
Fig. 4. Acute exposure group results. Mean thymine dimers (TT) for (a) from HPLC-tandem mass spectrometry (HPLC-MS/MS) expressed as TT/106 normal base pairs, (b) whole epidermis, (c) basal layer and (d) melanocytes from immunostaining. Data for (b), (c) and (d) are expressed as % of the 4 standard erythema doses (SED) positive control without sunscreen.
Table II. P-values for Bonferroni corrected post hoc comparisons of non-sunscreen (SS) treated control and SS treated sites, and generalised estimating equation (GEE) linear regressions for SS thickness in acute and repeated exposure groups. SED: standard erythema dose
The analysis of the HPLC-MS/MS data was restricted to TT CPD because this was the most frequent lesion in the acute group (Table I). The trends for all endpoints are very similar (Fig. 5). Unlike the acute study, there were no HPLC/MS-MS samples with zero values. There were also no zero values in the immunostained samples with 1 SED daily positive control exposures without sunscreen, and only 1 zero value (melanocytes) with the sunscreen at 0.75 mg/cm2. However, ≤ 4/8 of the other sunscreen treated samples had zero values. The highest numbers of zero values (overall epidermis, basal layer and melanocytes) were seen with 15 SED daily and the sunscreen at 1.3 and 2.0 mg/cm2. Zero values were included in the analyses. Considerable DNA damage is seen immediately after the 5th consecutive 1 SED SSR exposure without sunscreen vs. 0 SED (p ≤ 0.002) for all assays. ANOVA of all SSR treated sites/assays showed p ≤ 0.014. The Bonferroni-adjusted post hoc tests are shown in Table II. There was significantly less damage with 15 SED/day with sunscreen at 1.3 and 2.0 mg/cm2 than 1 SED/day with no sunscreen for all assays. There was also less damage with the sunscreen at 0.75 mg/cm2, but this difference was only significant for the HPLC-MS/MS assay. A comparison of 30 SED/day with the sunscreen at 2 mg/cm2 showed significantly less damage than 1 SED/day without sunscreen for all assays. ANOVA of the sunscreen-treated sites/assays alone showed no significant differences between any of the sunscreen treated sites (p = 0.054–0.437). How-ever, GEE analysis showed a significant linear trend for increasing protection with increasing sunscreen thickness with 15 SED/day except for melanocytes. A paired t-test comparing 15 and 30 SED with the sunscreen at 2.0 mg/cm2 showed less damage with 15 SED in all assays, but no difference was significant (p > 0.07).
Fig. 5. Repeat exposure group results. Mean thymine dimers (TT) for (a) from HPLC-tandem mass spectrometry (HPLC-MS/MS) expressed as TT/106 normal base pairs (b) whole epidermis, (c) basal layer and (d) melanocytes from immunostaining. Data for (b), (c) and (d) are expressed as % of the 4 standard erythema doses (SED) positive control without sunscreen.
High solar UVR doses can be achieved at tropical and sub-tropical latitudes where many fair-skinned Caucasians take their holidays. For example, exposures of 20–60 SED per day are possible in Brazil from 15–24°S, and 30 SED is achievable from 10.00–14.00 in mid-summer in São Paulo (24°S) (27). Sixty SED daily is possible in Southern Europe and 40–50 SED in Northern Europe (8). Considerable epidermal DNA damage (TT CPD) was measured in European holidaymakers after a week in March in Tenerife (28.3°N – comparable latitude to the holiday destination of Florida) (28), during which a mean cumulative dose of 57 SED over the week (range 21.0–115.0) was received by Danish holidaymakers (29), that was estimated to be 43% of their annual UVR burden. Furthermore, all the Danes had sunburn (7) despite the use of sunscreens. Thus, our experiments involved environmentally relevant exposures of 15 and 30 SED, and resulted in cumulative doses of 75 and 150 SED (~25 and ~50 MED) in the repeated exposure group.
The HPLC-MS/MS assay quantified overall epidermal TT CPD but the data also include some dermal lesions, the relative proportion of which will be very small (estimated < 5%) because the dermis (which was trimmed) is mainly an acellular tissue. The HPLC-MS/MS data were supported by immunostaining assessments of the epidermal location of CPD. Comparable trends and statistical outcomes were observed for all assays (see Table II) in both study groups. DNA protection was dependent on sunscreen application thickness in all assays except for melanocytes, but a linear trend was indicated. An acute exposure of 30 SED, with sunscreen at 1.3 and 2.0 mg/cm2, caused significantly less damage than 4 SED without sunscreen in all assays. However, there was no significant difference between DNA damage at 1.3 and 2.0 mg/cm2 application thicknesses in any of the assays. This, coupled with the zero values, may explain why there are more CPD with 2 mg/cm2 than 1.3 mg/cm2 in the HPLC/MS-MS assay (see Fig. 4a). Thirty SED plus sunscreen at 0.75 mg/cm2 also resulted in fewer TT CPD than 4 SED without protection. This difference was not significant in any assay, but shows less damage with 7.5 times the SSR control dose. Fig. 3b shows CPD hotspots with the sunscreen at 0.75 mg/cm2. This is not surprising because sunscreen films on the skin are composed of many different thicknesses, as formulations tend to fill valleys and leave peaks that are only partially protected (30).
The acute study also showed similar trends in the formation and protection of TT, TC and CT CPD (Table I). Cytosine containing CPD cause mutagenic C to T transitions in model systems (31) and skin tumours (32, 33). There was also a comparable induction of 6-4PP and their Dewar valence isomers on the various treatment sites. The respective photoproduct yields in the acute 4 SED positive controls agree with previous reports in skin exposed to UVB (280–315 nm) (21, 22) and SSR (34), which show fewer 6-4PP and their Dewar valence isomers than CPD, and that the Dewar TC derivatives are more frequent than the TT. Furthermore, we report here for the first time, the presence of Dewar isomers in human skin exposed in vivo to SSR. This confirms the major role of the solar UVA in the photoisomerization of 6-4PP (35–37). It should be noted that the relative frequencies of different types of DNA photoproducts in vitro depends on solar spectrum (latitude) (38). However, the UVB and UVA EEE of SSR and the Tenerife spectrum are very similar, but the spectrum that reaches the viable epidermis will be modified by a sunscreen’s optical properties.
Acute studies do not represent holiday exposure. Epidermal CPD accumulation in DNA occurs with daily sub-erythemal SSR exposure (39) because of slow nucleotide excision repair (NER). This is illustrated by the persistence of TT CPD in human skin in vivo with a half-life of 33.3h (18). An acute control exposure of 4 SED (~1.3 MED) gave a mean ± SD of 30.1± 14.8 TT CPD/106 bases whereas 5 daily 1 SED (~0.3 MED) induced 16.8± 4.9 TT CPD/106 bases. The lower value (56%) with the higher cumulative dose is indicative of NER during the 5-day study.
The repeated exposure studies showed that protection was significantly dependent on sunscreen application thicknesses for all assays except melanocytes. Daily high dose exposure with sunscreen at 1.3 mg/cm2 and 2.0 mg/cm2 resulted in significantly less DNA damage than 1 SED/day without protection in all assays. This was also the case for sunscreen at 0.75 mg/cm2 with the quantitative HPLC-MS/MS assay but not for the immunostaining endpoints. The lack of detectable lesions by immunostaining in some cases with the sunscreen at 1.3 and 2.0 mg/cm2 is also indicative of a high level of protection.
The SPF is a function of sunscreen application thickness (9, 40), which we also show for DNA protection, for the first time, under acute and repeated SSR exposure conditions. A recent study of holidaymakers in Egypt (9) showed a mean sunscreen application thickness of 0.79 mg/cm2 (labelled average SPF=15 with estimated real SPF=3), which is very similar to our value of 0.75 mg/cm2. This typical “real life” application thickness in the acute exposure group showed no significant difference in TT CPD, in any assay, compared with unprotected skin (see Fig. 4). The SSR dose was 7.5 times higher in the acute study for protected skin (at 0.75 mg/cm2) compared with unprotected skin, which indicates a DNA protection factor (DNA-PF) of ≥ 7.5 (30/4 SED). A comparable analysis with the immunological data indicates a DNA-PF of ≥ 15 (15/1 SED) in the repeated exposure group. However, the difference was significant with the HPLC-MS/MS assay with 40% TT CPD on sunscreen treated sites compared with 1 unprotected SED (see Fig. 5a), giving a crude estimated DNA-PF of 37.5 (15/0.4). Overall, our data demonstrate considerable DNA protection by a sunscreen even under typical conditions of application, intense tropical holiday insolation and, importantly, repeated SSR exposure.
“Dark” CPD have been recently described in a murine model and in vitro after UVA radiation. These lesions are formed post-irradiation, with a peak of 3–4 h after exposure (41, 42). Vitamin E (α-tocopherol) inhibited the formation of “dark” CPD in the in vitro studies. We have reported the presence of “dark” CPD in human skin in vivo after SSR exposure (Fajuyigbe et al, manuscript under preparation). The sunscreen contained a vitamin E based antioxidant (25). It is possible that some of its CPD protective effects were mediated via the inhibition of “dark” CPD. However, the taking of the biopsies immediately after exposure (or the last exposure in the repeated exposure group) was not designed to assess the effect of the sunscreen on “dark” CPD.
A recent study has reported the effect of the same sunscreen at 2 mg/cm2 after individualised MED-based SSR exposures in skin type I/II volunteers (43). Analyses by HPLC-MS/MS were done on epidermal DNA samples derived from suction blisters. TT CPD (and CT and TC CPD) were detected after 2 MED (~4–6 SED (21)) in the same rank order and at comparable quantities with our 4 SED control data. However, no lesions were detected after 15 MED (~30–45 SED) in the presence of sunscreen. 15 MED though an SPF = 64 sunscreen is 0.25 MED (0.5–0.75 SED). The blisters were taken at ~4 h after exposure, which will also have allowed time for maximal “dark” CPD to be formed and very limited repair of “light” CPD (18).
The introduction reports that sunscreen use can significantly inhibit melanoma, SCC but not BCC. However, the sunscreen protection against skin cancer offered by sunscreen is very modest; for example rate ratio of 0.62 (95% CI 0.38–0.99) for SCC with long-term follow up (11) and hazard ratio of 0.50 (95% CI 0.24–1.02) for melanoma in the Australian study population (13); in effect the sunscreen offered “skin cancer protection factors” of ≤ 2 with a labelled SPF of 16 but in practice likely to have been considerably lower (44). It is well established that the incidence of all types of skin cancer is much lower in black skin compared to white (45). For example, the incidence of melanoma is ~30 times greater in white vs. black Americans (46). The DNA-PF that constitutive melanin (on sun protected skin) affords against SSR-induced CPD has been determined by comparing black (type VI) and white (types I/II) skins (47). The DNA-PF was dependent on epidermal location, ranging from 5.0 (95% CI 4.5–5.5) in the upper epidermis to 59.0 (95% CI 24–110) in the basal layer that has the greatest concentration of melanin. This suggests that high SPF sunscreens are necessary to have a major impact on skin cancers, especially those that originate in the basal layer.
The study strengths are the assessment of the sunscreen under conditions that simulate intense real life holiday solar exposure and typical application thickness of sunscreens. The comparable outcomes with quantitative HPLC-MS/MS and semi-quantitative immunostaining add robustness to our conclusions. Protection is also shown in the basal layer that contains keratinocyte stem cells and melanocytes from which skin cancers can arise. The trends with the melanocytes were similar to the other endpoints but with higher p-values; almost certainly due to their relatively small number compared to the other endpoints. The weakness of the study is the relatively small numbers of participants, though, as shown in Table II, most of the comparisons show a high level of significance. Furthermore, this study was not designed to determine sunscreen DNA-PF, which is an important parameter for future research. Prevention of CPD provides a mechanistic basis for the ability of sunscreens to inhibit SCC, and possibly melanoma (48). Our data support the role of regular sunscreen use to reduce the risk of skin cancer and the application of very high SPF sunscreens, even if they are typically used at ~1/3 of the thickness required for SPF labelling.
The research was funded by Pierre Fabre Dermo-Cosmétique, and was supported by the National Institute for Health Research (NIHR) Clinical Research Facility at Guy’s & St Thomas’ NHS Foundation Trust and NIHR Biomedical Research Centre based
at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
We thank Drs Angela Tewari, Mieran Sethi and Ms Hemawtee Sreenebus for the taking of the biopsies. We thank all our volunteers. We thank Dr Victoria Cornelius, Senior Lecturer in Clinical Trial Statistics, Imperial College London, for the statistical analyses. We thank the Pierre Fabre Clinical Research team for the support with the study management and monitoring (Isabelle Martin), quality control (Christophe Chamard), statistical and data management (Christophe Lauze and Chantal Sovran), Victor Georgescu and Ulrike Sattler from the Avène medical team for the input in the protocol and manuscript review, and Lara Claude-Braeuer for the preparation of the clinical report.
FB, GJ, EQ, CM and ABR are employees of Pierre Fabre Dermo-Cosmétique. ARY has received travel grants from Pierre Fabre Dermo-Cosmétique.


 Click to show fullsize
Click to show fullsize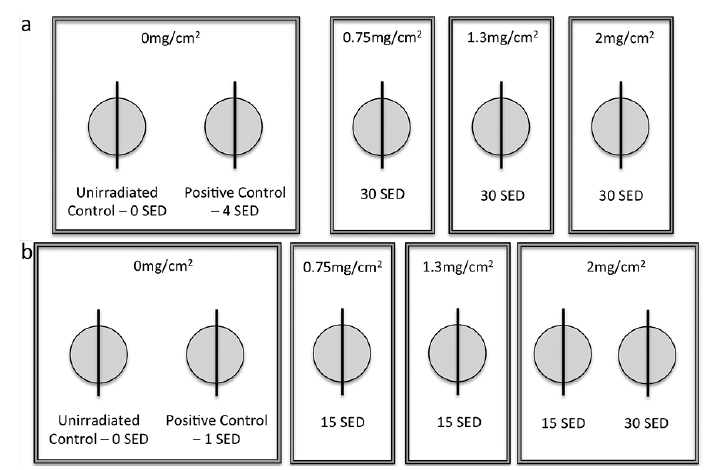 Click to show fullsize
Click to show fullsize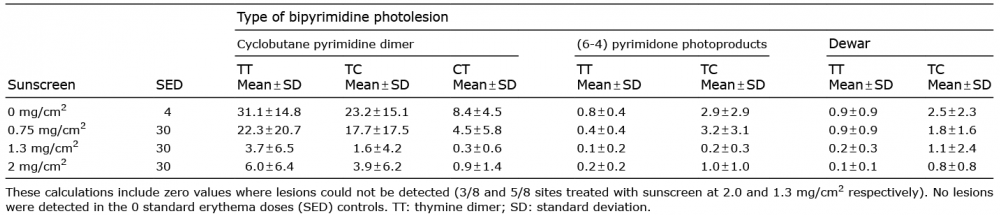 Click to show fullsize
Click to show fullsize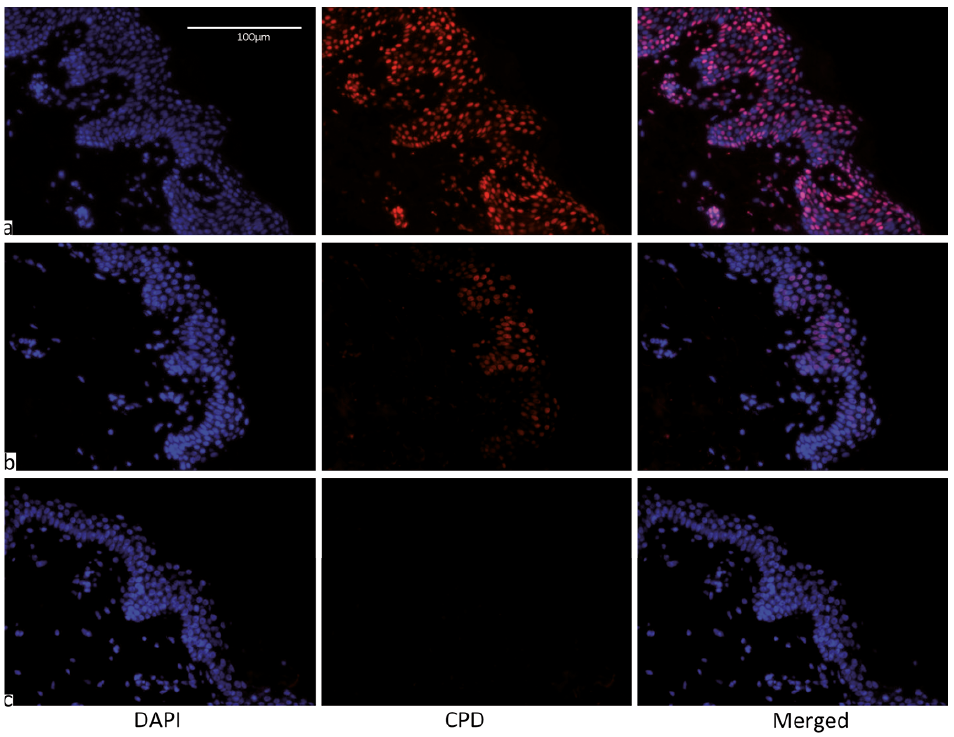 Click to show fullsize
Click to show fullsize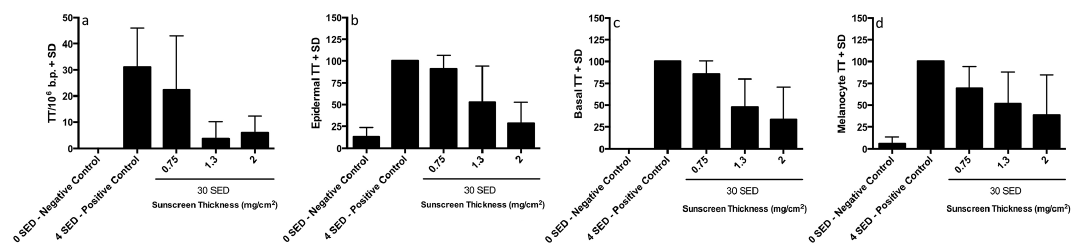 Click to show fullsize
Click to show fullsize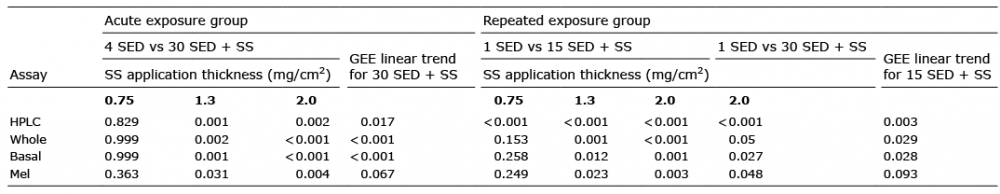 Click to show fullsize
Click to show fullsize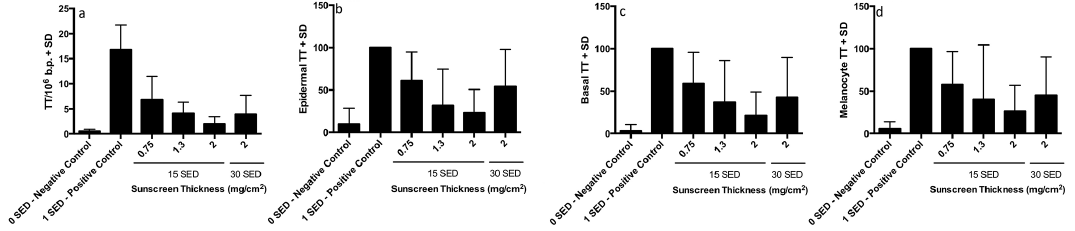 Click to show fullsize
Click to show fullsize