


1Dermatology and Venereology Section, Department of Medicine Solna, Karolinska Institutet, 2Dermatology Unit, Karolinska University Hospital, Stockholm, Sweden, and 3Dermatology Unit, Wuhan No.1 Hospital, Wuhan, China
Psoriasis is a common immune-mediated disease resulting from altered cross-talk between keratinocytes and immune cells. Previous transcriptomic studies have identified thousands of deregulated genes in psoriasis skin; however, the transcriptomic changes confined to the epidermal compartment remained poorly characterized. The aim of this study was to characterize the transcriptomic landscape of psoriatic keratinocytes, using sorted CD45neg epidermal cells. Genes with functions in innate immunity, type I interferon response, cell cycle and keratinization were enriched among deregulated genes in psoriatic keratinocytes. Gene set enrichment analysis indicated the dominance of interleukin (IL)-22/IL-17A signatures in the epidermal psoriasis-signature. A set of deregulated genes overlapped with psoriasis-associated genetic regions, suggesting that genetic variations affecting gene expression in keratinocytes contribute to susceptibility to psoriasis. Several psoriasis-susceptibility genes, which were previously believed to be expressed preferentially or exclusively in immune cells, were identified as having altered expression in psoriatic keratinocytes. These results highlight the role of keratinocytes in the pathogenesis of psoriasis, and indicate that both genetic factors and an inflammatory microenvironment contribute to epidermal alterations in psoriasis.
Key words: psoriasis; keratinocyte; transcriptome; cytokine; epidermis.
Accepted Oct 15, 2018; Epub ahead of print Oct 15, 2018
Acta Derm Venereol 2019; 99: XX–XX.
Corr: Enikö Sonkoly, Dermatology and Venereology Section, Department of Medicine Solna, Karolinska Institutet, CMM L8:02, SE-171 76 Stockholm, Sweden. E-mail: eniko.sonkoly@ki.se
Psoriasis is a common inflammatory skin disease resulting from an interplay of skin cells (keratinocytes) and immune cells. While more than 1,000 genes have altered expression in psoriasis skin, the contribution of keratinocytes to these changes is poorly characterized. This study found that keratinocytes from psoriasis skin display marked changes in gene expression, associated with proliferation, differentiation and genes induced by inflammatory mediators. Part of the identified genes overlap with genetic regions associated with susceptibility to psoriasis. These results suggest that keratinocytes in psoriasis have both intrinsic (genetic) and extrinsic (inflammation-induced) changes, and highlight the role of keratinocytes in psoriasis.
Psoriasis is a common chronic inflammatory skin disease that affects > 125 million people worldwide (1). It is a multifactorial disease (2), with both genetic and environmental factors contributing to its development. Genome-wide association studies have identified more than 40 psoriasis-associated single-nucleotide polymorphisms (SNPs) (3) located near to genes linked to innate and adaptive immunity as well as keratinocyte differentiation (4).
The characteristic skin lesions in psoriasis are ery-thematous scaly plaques, characterized by epidermal hyperplasia and epidermal and dermal infiltration of immune cells (2). Psoriatic skin inflammation is thought to develop as a result of abnormal communication between infiltrating immune cells and activated keratinocytes. Th17 cells and their secreted cytokines, interleukin (IL)-17, and IL-22, in synergy with interferon (IFN)-γ and tumour necrosis factor alpha (TNF-α), have been identified as central players in the pathogenesis (2, 5). Keratinocytes, which were previously thought of as passive bystanders in the inflammatory process, play an active role both in the initiation and maintenance of psoriatic skin inflammation, by secreting chemokines, cytokines, and antimicrobial peptides, which lead to further chemoattraction and activation of immune cells in the skin, amplifying the inflammatory process (5). Moreover, psoriatic keratinocytes follow an altered differentiation programme and have an increased proliferation rate.
Previous transcriptomic studies on full-depth skin biopsies have demonstrated profoundly altered gene expression in psoriasis plaques, with thousands of differentially expressed genes (DEGs) (6) compared with healthy skin. Gene expression changes, although with a lower number of DEGs, have been detected even in the non-lesional, healthy-looking skin of patients with psoriasis, suggesting a “pre-psoriatic” state of non-lesional skin. However, most transcriptomic studies in psoriasis have utilized full-depth skin biopsies (6–9). Although several attempts have been made to characterize transcriptomic changes confined to keratinocytes, the contribution of the epidermal compartment to the total changes in psoriasis skin is poorly characterized (10–12), is poorly characterized (10–12), and these previous studies have some limitations: microdissected epidermal tissue contains a mix of different cell types; the epidermis contains a very large number of immune cells, such as CD4+ and CD8+ T cells, neutrophil granulocytes, macrophages, and dendritic cells (DCs) (5, 13), which are present in higher numbers in psoriasis compared with healthy skin. Moreover, cell type-specific expression changes can be masked by expression in other cell types when studying bulk tissue with different cell types. The use of cultured keratinocytes overcomes this problem; however, these display overt phenotypic and transcriptional changes especially in genes related to proliferation and differentiation, potentially masking differences in the tissue.
This study explored the transcriptomic landscape of psoriatic keratinocytes, using sorted CD45neg epidermal cells, in which immune cell transcriptomes are excluded. Using this approach, keratinocyte-specific gene expression changes in lesional and non-lesional psoriasis skin were identified.
Punch (4 mm) biopsies were collected from 9 healthy donors (5 women and 4 men, age range 26–55 years) and from 9 lesional and non-lesional skin of patients with chronic plaque psoriasis (3 women and 6 men, age range 24–69 years) at the Department of Dermatology, Karolinska University Hospital, Stockholm, Sweden and at the Swedish Psoriasis Association (Psoriasisföreningen). Biopsies from non-lesional skin were taken at least 10 cm from the nearest psoriasis plaque. Patients had not received systemic immunosuppressive treatment or ultraviolet B (UV-B)/psoralen plus ultraviolet A (PUVA) treatment for at least 4 weeks, or topical therapy for at least 2 weeks before skin biopsy. Written informed consent was obtained from all patients prior to biopsy. The study was approved by the regional ethics committee and performed according to the principles of the Declaration of Helsinki.
Skin biopsies were incubated in dispase (5 U/ml) overnight at 4°C, and epidermis and dermis were separated. The epidermis was cut into smaller pieces, digested with trypsin/EDTA, and incubated for 15 min at 37°C. CD45neg cells were sorted by depleting CD45pos cells using CD45-microbeads in MACS MS magnetic columns according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany).
Total RNA from sorted CD45neg epidermal cells was extracted using TRIzol (Life Technologies, Carlsbad, CA, USA). Microarray analysis of gene expression was performed using the platform Human Transcriptome Array 2.0 (Affymetrix, Santa Clara, CA, USA), containing > 6.0 million probes. Limma (Linear Models for Microarray Data), a Bioconductor package, was used to identify DEGs. The Benjamini-Hochberg approach was used for multiple hypothesis correction. Protein-coding genes were filtered according to the following threshold criteria: linear fold-change <0.67 and >1.5, and false discovery rate (FDR)<0.05.
The online tool Enrichr (14, 15) was used to perform Gene Ontology Biological Processes (GO-BP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis on the whole list of 2,365 significantly DEGs (fold-change < 0.67 and > 1.5, FDR< 0.05) among the psoriasis lesion (PP) and healthy (H) samples comparison. KEGG 2016 and GO-BP 2017b information were retrieved to determine the most relevant biological patterns and then plotted according to their significance (–log10(p-value)).
A total of 36 lists of DEGs in cytokine-treated keratinocytes monolayer and 3D reconstructed epidermis (see Table SIII) were obtained from the Gene Expression Omnibus (GEO) database (http://www. ncbi.nlm.nih.gov/geo/) and were used to perform Gene Set Enrichment Analysis (GSEA) to identify cytokine-responsive gene signatures in keratinocytes that are enriched in the CD45neg epidermal cells from psoriasis lesional skin. Genes up-regulated by cytokines treatments with fold-change > 1.5 and nominal p-value < 0.05 were considered for the enrichment analysis, carried out using GSEA software (powered by Broad Institute). For these analyses the expression of the genes in our list was normalized and reverse log2 transformed.
SNPs associated with psoriasis susceptibility loci (PSORS) with relative chromosome coordinates were extracted from 4 publications: (3, 4, 16, 17). Knowing the exact location of the PSORS-associated SNPs, the’ genomic coordinates of differentially expressed genes were overlapped with a range of 500 kbp (250 kbp up-stream and 250 kbp down-stream of the location of the SNP) through the R based Bioconductor package “GenomicRanges” (16) (https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html). If the gene coordinates were entirely within the 500 kbp range of the SNP, this was classed as a complete overlap; if they were partially overlapping it was classed a partial overlap. For a full list of the identified genes co-localized in the SNPs PSORS-associated region, see Table SIV.
MetaCore™ (Thomson Reuters, New York, NY, USA) software, using published predicted and validated data, was used to determine the transcription factor binding motifs among the 2,365 DEGs in psoriasis lesion (PP) vs. healthy controls (H).
Total RNA was reverse transcribed using the RevertAid™ First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, USA). TaqMan® PrimeTime predesigned qPCR primers for genes validation were purchased from Integrated DNA Technologies (IDT, Coralville, USA). Quantitative real-time PCR (qRT-PCR) was carried out as described by the manufacturer (Applied Biosystems, Foster City, CA, USA) and the values were normalized to the expression of the housekeeping gene 18S.
Normal human epidermal keratinocytes (HEKa) (Thermo Fisher Scientific, Catalog# C0055C) were treated with recombinant Epigen (rEPGN) protein (Novus-Biologicals, Catalog# NBP2-34987) at 3 increasing concentrations (50, 100, 200 ng/ml). Proliferation assay was performed with IncuCyte ZOOM® Live-Cell Analysis System and area among the proliferating cells was analysed with the IncuCyte ZOOM® software.
To identify transcriptomic changes in psoriatic keratinocytes, skin biopsies were obtained from lesional (PP) and non-lesional (PN) skin of 9 patients with psoriasis, and from healthy skin of 9 volunteers (H). Keratinocytes were sorted out from the epidermis by negative sorting for the immune cell marker CD45, using magnetic cell sorting (MACS). Next, transcriptomic analysis was performed on this purified cell population. The purity of the sorted CD45neg cells was confirmed by the microarray expression data showing high expression of keratinocyte-specific markers (e.g. K5, S100A7) and the absence of immune cell-specific markers (e.g. CD3, IL-8, CXCL9) in the CD45neg cell population (Fig. S1).
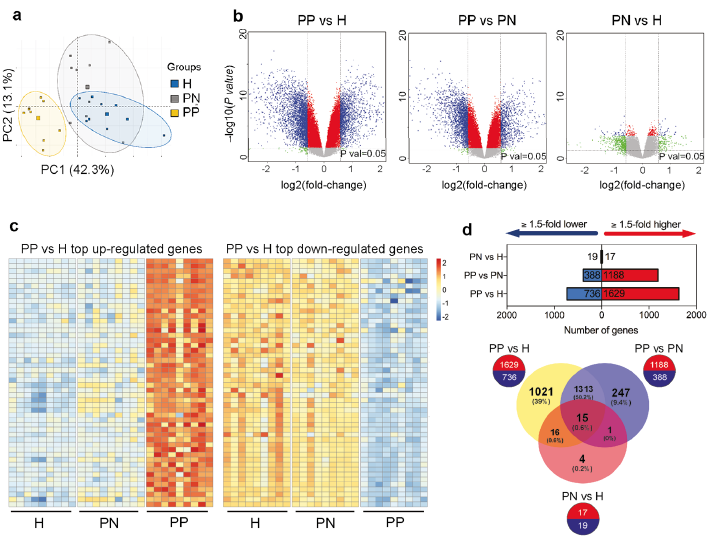
Principal component analysis (PCA) revealed clear segregation of psoriasis lesional keratinocytes from psoriasis non-lesional or healthy samples (Fig. 1a). DEGs were identified based on 3 comparisons: (i) PP (psoriasis lesional) vs. H (healthy), (ii) PP vs. PN (psoriasis non-lesional), and (iii) PN vs. H. Comparison of the epidermal transcriptome of PP with that of H identified 2,365 DEGs, with FDR < 0.05, and fold change > 1.5, of which 1,629 genes were up-regulated and 736 were down-regulated (Fig. 1c–d; Table SIa). A paired comparison of the keratinocyte transcriptome of psoriatic lesional and non-lesional skin (PP vs. PN) revealed 1,576 DEGs, out of which 1,188 were up-regulated and 388 down-regulated (Fig. 1d; Fig. S2a; Table SIb); the lower number of DEGs in the PP vs. PN comparison indicates that the transcriptomic profile of PN keratinocytes is more similar to PP than that of H keratinocytes, suggesting a “pre-psoriatic” state in the non-lesional keratinocytes. The PN vs. H comparison revealed 36 DEGs with >1.5-fold change (Fig. 1d; Fig. S2b; Table SIc). Thirty-two out of these 36 genes (88.9%) were also differentially expressed in the other comparisons (Fig. 1d).
Fig. 1. Transcriptomic changes in keratinocytes in psoriasis skin. (a) Principal component analysis of the transcriptome of sorted CD45neg epidermal cells from lesional (keratinocytes from psoriasis lesional skin (PP), yellow squares) and non-lesional (keratinocytes from psoriasis non-lesional skin (PN), green squares) skin of psoriasis patients and skin from healthy controls (keratinocytes from healthy skin (H), blue squares). (b) Volcano plots plotting the log2 fold-change and the nominal p-value (grey horizontal line) and the adjusted p-value for all the transcripts detected by the Affymetrix platform in each comparison. Blue colour indicates transcripts with fold-change (FC) <0.67 while red colour those with FC >1.5 (c). Heatmaps showing the top 50 up-regulated (left) and top 50 down-regulated (right) genes in psoriatic keratinocytes (PP vs. H), FDR <0.05 and FC >1.5 or FC <0.67 for the up- and down-regulated genes, respectively. (d) Graphs representing the numbers of significant genes (FDR<0.05) with a >1.5-fold increase (red) or decrease (blue) in expression levels between the indicated groups; the Venn diagram shows the extent of overlap among differentially expressed genes with >1.5-fold increase/decrease from each comparison.
In order to identify biological functions/pathways enriched among the DEGs in psoriatic keratinocytes, GO (Gene Ontology)/KEGG (Kyoto Encyclopedia of Genes and Genomes)-based enrichment analysis was performed on DEGs on psoriatic keratinocytes (PP vs. H, FC > 1.5 and < 0.67, p-value < 0.05) (Fig. 2). Common relevant terms identified among up- and down-regulated genes were “keratinocyte differentiation” (GO:0030216, FDR=1.03×10–3 for up-regulated genes and FDR=1.20×10–9 for down-regulated genes) and “keratinocyte development” (GO:0003334, FDR=8.56×10–4 for up-regulated and FDR=6.87×10–10 for down-regulated genes). The most significantly enriched terms among the up-regulated genes in keratinocytes of psoriasis lesions (PP vs. H) included cellular functions related to cell proliferation, such as “DNA-dependent DNA replication” (GO:0006261, FDR=1.21×10–11), immune and inflammatory processes, such as the “type I interferon signaling pathway” (GO:0060337, FDR=3.00×10–3) and “NIK/NF-κB signaling” (GO:0038061, FDR=2.19×10–3). For the down-regulated genes in PP vs. H comparison, some of the top significant processes were related to keratinocyte development/differentiation e.g. “skin epidermis development” (GO:0098773, FDR=1.18×10–6). In addition, significant KEGG pathways were associated with apoptosis, cellular tight junction and TGF-β signalling (Fig. 2; a list of genes in the identified significantly enriched GO and KEGG pathways is shown in Table SII). Cytoscape network analysis confirmed significant enrichment of biological processes (GO categories) related to keratinocyte differentiation, cell cycle, type I interferon response and epidermis development among the DEGs; moreover, genes involved in cytokine response (GO:0034097, FDR=3.46×10–8) and cornification (GO:0070268, FDR=1.91×10–6) were significantly enriched (Fig. S3). In the PP/PN comparison, GO (gene ontology)/KEGG-based enrichment analysis showed similar results as in the PP/H comparison, showing enrichment of keratinocyte development and differentiation among the top significant biological processes, cell cycle and DNA replication among the top significant pathways (data not shown).
Fig. 2. The keratinocyte transcriptome of psoriasis lesions is enriched for genes related to cell cycle, innate immunity, epidermis/keratinocyte development and differentiation, and DNA repair/replication. Genes deregulated in psoriatic keratinocytes (keratinocytes from psoriasis lesional skin (PP),vs. keratinocytes from healthy skin (H)) were analysed using Enrichr. The top 10 most relevant biological processes (gene ontology (GO), upper graph) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways are sorted according to the adjusted p-value (calculated using the Benjamini-Hochberg method for correction for multiple testing).
Next, this study aimed to identify the upstream regulators of the genes differentially expressed in psoriatic keratinocytes. Since psoriatic keratinocytes are exposed to an inflammatory cytokine milieu, altered expression of part of the identified genes can be driven by the altered cytokine environment in psoriasis lesions. To determine whether the identified up-regulated DEGs in psoriatic keratinocytes are disproportionally induced by one or more cytokines or growth factors, GSEA was performed using 36 set of genes that were regulated upon cytokine/growth factor treatment of cultured primary keratinocytes or epidermal equivalents (Table SIII), available on the GEO database. Results of GSEA demonstrated that genes induced by IL-22 and IL-17A treatment in primary monolayer keratinocytes showed the most significant enrichment among the identified up-regulated DEGs in psoriatic keratinocytes, followed by IL-20, IL-24 and IL-1α (Fig. 3a and Fig. S4). Moreover, a significant positive enrichment was observed for genes up-regulated by epidermal growth factor (EGF) in keratinocytes (Fig. 3a). In contrast, genes induced by the Th2 cytokine IL-13 or by TGF-α were overrepresented at the bottom of the ranked list of genes from the expression dataset, i.e. they were repressed in psoriatic keratinocytes. These results provide evidence for the dominance of an IL-22/IL-17A signature in psoriatic keratinocytes, with contribution of the IL-20 subfamily, the IL-1/IL-36 family, TNF-α and IFN-γ. In total, 770 (47.3%) up-regulated genes in psoriatic keratinocytes did not overlap with any of the cytokine-induced signatures (Fig. 3b), indicating that there can be intrinsic alterations in psoriasis epidermis that are not dependent on the inflammatory cytokine milieu. Interestingly, considering the number of cytokine-induced genes among PP vs. H up-regulated DEGs, IFN-γ-induced genes represent the highest proportion (21.5%) followed by IL-22-induced genes, which constitute 16% of up-regulated genes in psoriatic keratinocytes (Fig. 3C). Nine percent of the PP vs. H up-regulated genes overlap with IL-17A-induced genes; thus, the IL-17A-signature is represented by fewer, but more significantly regulated, genes (Fig. 3a) compared with the IFN-γ signature.
Fig. 3. The keratinocyte transcriptome of psoriasis is dominated by IL-22/IL-17A signatures. (a) Gene Set Enrichment Analysis (GSEA) was performed to evaluate enrichment of genes induced by cytokines/growth factors in cultured keratinocytes, among up-regulated DEGs in psoriatic keratinocytes (keratinocytes from psoriasis lesional skin (PP),vs. keratinocytes from healthy skin (H)). Thirty-six gene sets of cytokine/growth factor-regulated genes in keratinocytes were obtained from Gene Expression Omnibus (GEO). Genes with fold change >1.5 and p-value < 0.05 were considered differentially expressed. Red bars denote positive enrichment (i.e. genes disproportionally induced), while blue bars denote negative enrichment (i.e. genes disproportionally repressed). (b) Pie chart showing the number of up-regulated DEGs in psoriatic keratinocytes (PP vs. H) which are (dark green, “cytokine/growth factor-dependent”) or are not (light green, “cytokine/growth factor-independent”) overlapping with any of the 36 gene sets of cytokine-induced genes in keratinocytes. (c) Pie charts representing the proportion of genes significantly induced by each cytokine/growth factor among all the genes induced in PP vs. H comparison.
Next, we aimed to explore the contribution of genetic factors to differential gene expression in psoriatic keratinocytes. To this end, we analysed the genomic localization of the identified DEGs in psoriatic keratinocytes (PP vs. H) and their potential overlap with regions in the proximity of SNPs associated with psoriasis susceptibility. Psoriasis-associated SNPs were identified from 4 genome-wide association studies (GWAS) (3, 4, 17, 18). We found that 107 out of 2,365 DEGs in the lesional (PP) vs. healthy (H) comparison were confined to a region of 250 kbp up- and down-stream of the psoriasis-associated SNPs (Fig. 4a, d); 80 out of 1,576 DEGs in lesional (PP) vs. non-lesional (PN) groups (Fig. 4b, e); and 2 out of 36 DEGs in non-lesional (PN) vs. healthy (H) groups comparison were localized in the proximity of psoriasis-associated SNPs (Table SIV). Analysis of the chromosomal distribution of DEGs in psoriatic keratinocytes (PP vs. H) localized in the proximity of psoriasis-associated SNPs showed that nearly one-third of them (29/90 in total) are localized on chromosome 1 (Fig. 4c). These included genes within the epidermal differentiation complex (EDC) associated with keratinocyte terminal differentiation and skin barrier, such as late cornified envelop (LCE) genes, SPRR4, KPRP, FLG2, IVL and C1orf68 (19). In our data, C1orf68 (xp32) was the most down-regulated protein-coding gene, which is also one of the closest to the PSORS4-associated SNPs on chromosome 1q21. In addition to known psoriasis-susceptibility genes, our analysis revealed genes with differential expression in psoriatic keratinocytes that are near to psoriasis-associated SNPs, but have not been functionally characterized in the context of psoriasis (Table SIV).
Fig. 4. Genes differentially expressed in psoriatic keratinocytes overlap with regions in proximity to psoriasis-associated single-nucleotide polymorphisms (SNPs). (a) Psoriasis lesional skin (PP) vs. healthy skin (H) differentially expressed genes (DEGs), and (b) PP vs. psoriasis non-lesional skin (PN) DEGs, in the proximity of SNPs associated with psoriasis, based on previous genome-wide association studies (GWAS). Bars indicate log2 transformed fold change (log2FC, upper graph) and distance from the psoriasis-associated SNP in kbp (lower graph). (c) Bar graph illustrating the chromosomal distribution of DEGs (fold-change <0.67 or >1.5, FDR<0.05) in the PP vs. H comparison. Red sections in each bar represent the number of genes overlapping with regions at <250 kbp distance from psoriasis-associated SNPs identified from GWAS. Fifteen well-established psoriasis susceptibility (PSORS) regions are indicated above the bars. (d, e) Heatmap showing expression of DEGs in psoriatic keratinocytes ((d): PP vs. H, (e) PP vs. PN comparison) overlapping with regions at <250 kbp distance from psoriasis-associated SNPs identified from GWAS. FC: fold change.
To determine if functionally clustered genes were coordinately controlled by common transcription factors, MetaCore™ analysis of the 2,365 DEGs in the PP vs. H comparison was performed. Results of the analysis revealed significantly enriched binding sites for 338 transcription factors. Out of these, 54 transcription factors had significantly altered mRNA levels (17 were significantly upregulated and 37 were downregulated) in psoriatic keratinocytes (Fig. 5 and Table SV). These include transcription factors known to regulate epidermal differentiation and inflammation, among others several subunits of the AP-1, as well as the NF-κB transcription factors, SOX4, KLF4, GATA3, members of the E2F family, STAT1, STAT2 and STAT3, but also transcription factors previously less characterized in the psoriasis context, such as TRPS1 (Transcriptional Repressor GATA Binding 1), HEY2 (Hes Related Family BHLH Transcription Factor With YRPW Motif 2) and PAX3 (Paired Box 3). Transcription factors previously suggested to function in a T-cell intrinsic manner, such as FOXO1, were also deregulated in psoriatic keratinocytes (Fig. 5 and Table SV). It is noteworthy that several of the differentially expressed transcription factors (e.g. SOX4, KLF4, AP1, IRF4, STAT2/3) are located in previously characterized psoriasis susceptibility regions, thereby representing a link between genetic susceptibility to the disease and molecular alterations in psoriasis skin. Overall, our results have identified a set of transcription factors that can act as upstream regulators of DEGs in psoriatic keratinocytes.
Fig. 5. Over-represented transcription factor binding sites in genes differentially expressed in psoriatic keratinocytes. (a) Transcription factors with overrepresented binding sites among differentially expressed genes (DEGs) in psoriatic keratinocytes (psoriasis lesional skin (PP) vs. healthy skin (H)), identified by MetaCore analysis. Only transcription factors regulating at least 10 target genes are shown. Arrows indicate selected transcription factors previously identified in psoriasis, and different colours indicate different subfamilies or subunits of transcription factors.
To validate the results of the transcriptomic profiling we performed qRT-PCR for selected transcripts that have not been functionally characterized in psoriasis. qRT-PCR analysis confirmed the significant up-regulation of the type I interferon response genes IFI44, IFI44L and DDX60 in epidermal cells of psoriasis lesional, but not in non-lesional, keratinocytes (Fig. 6a, b and c). TNIP3, a negative regulator of NF-κB signalling (20), was nearly undetectable in keratinocytes of healthy and non-lesional skin, while it was induced in psoriatic lesional keratinocytes (Fig. 6d). NR4A3, a transcription factor, is one the genes identified to be significantly down-regulated in keratinocytes of non-lesional (PN) psoriasis skin samples compared with healthy keratinocytes. qRT-PCR confirmed its downregulation (0.51-fold, p < 0.05) in non-lesional psoriatic keratinocytes, and its downregulation was even more pronounced (0.23-fold, p < 0.001) in keratinocytes of psoriasis skin lesions (Fig. 6e). The epidermal growth factor epithelial mitogen (Epigen, EPGN) was undetected in healthy and non-lesional psoriatic keratinocytes, but was upregulated in psoriasis lesional keratinocytes (Fig. 6f). EPGN is a ligand of the epidermal growth factor receptor (EGFR), and has not been functionally characterized in psoriasis. To investigate whether EPGN affects the proliferation of keratinocytes, we treated normal human epidermal keratinocytes (HEKa) with Epigen recombinant protein (rEPGN) and assessed their proliferation rate using IncuCyte. Our results showed that EPGN significantly increased keratinocyte proliferation (Fig. 6g), suggesting that its increased expression in psoriasis may contribute to hyperproliferation of keratinocytes in psoriasis plaques.
Fig. 6. Quantitative real-time PCR (qRT-PCR) analysis of (a) IFI44, (b) IFI44L, (c) DDX60, (d) TNIP3, (e) NR4A3 and (f) EPGN in CD45neg epidermal cells from lesional (PP) and non-lesional (PN) skin of patients with psoriasis, and healthy skin of control subjects (H). Each data point represents samples from individual donors. Data were statistically analysed with Mann–Whitney test for unpaired samples in PP vs. H and PN vs H group comparison and with Wilcoxon test for paired samples in PP vs. PN group comparison. (g) IncuCyte analysis of keratinocyte proliferation upon recombinant epigen (rEPGN) treatment for 24 h. Data were analysed with 2-way analysis of variance (ANOVA). *p < 0.05, **p < 0.01, ***p < 0.001.
This study characterized the keratinocyte-specific transcriptomic signature of psoriatic lesional and non-lesional skin. Using magnetic sorting of CD45neg cells from the epidermis, a cell population that consists mainly of keratinocytes was obtained and used for transcriptomic profiling. Our approach differs from that of previous transcriptomic analyses of psoriasis skin utilizing full-depth skin biopsies (6–9) or epidermal tissue obtained by laser capture microdissection (12). Although dispase treatment and magnetic cell sorting may have affected the expression of some of the genes, a strength of our approach is that we have obtained a purer cell population and excluded not only the signature of dermal cells (infiltrating T cells, dendritic cells, fibroblasts, endothelial cells, etc.), but also that of intraepidermal immune cells (e.g. CD4+ and CD8+ T cells, neutrophil granulocytes, Langerhans and dendritic cells), which are known to be present in higher numbers in psoriasis skin (13).
Altered expression of more than 2,300 genes was detected in keratinocytes from psoriasis lesions compared with healthy controls. Among upregulated genes, functions related to cell proliferation, innate immunity and inflammatory responses were enriched, consistent with previous full-depth skin transcriptome studies (8, 9). Upregulation of proliferation-related genes is in line with epidermal thickening in psoriasis plaques. One of the previously uncharacterized transcripts in psoriasis was EPGN, the latest member of the EGF family discovered (21). We show that EPGN promotes keratinocyte proliferation and its induction may thus contribute to keratinocyte hyperproliferation in psoriasis.
One of the pathways enriched among upregulated genes was type I interferon response and viral infections (22). While IFN-α has been shown to be involved in the initiation of psoriasis (13), the upregulation of type I interferon signature also suggests an activation of the pathway in the keratinocytes of established plaques. Among inflammatory pathways, the NF-κB pathway was enriched, consistent with previous studies; notably, genes previously assigned to immune cells were identified, such as TNIP3, also called ABIN-3 (A20-Binding Inhibitor of NF-Kappa-B Activation 3), an IL-17-response gene (23), which was previously assigned to γδ T-cells (6), and identified here as a transcript overexpressed in keratinocytes (20). Among down-regulated genes the most enriched functions were linked to epidermis development/differentiation, reflecting the altered keratinocyte differentiation in psoriasis.
GSEA using cytokine-induced gene signatures in cultured keratinocytes (6) showed that IL-22- and IL-17A- signatures were dominant in the transcriptomic signature of psoriatic lesional keratinocytes, in line with the current immunopathological model of psoriasis as an IL-17/IL-22-driven disease. Notably, also the IFN-γ signature was enriched and, considering the number of DEGs, this was the most prominent cytokine-induced gene signature. Moreover, enrichment of an EGF signature was found, consistent with the hyperproliferative state of psoriatic keratinocytes. More than half of the upregulated genes in psoriasis lesions were not overlapping with any of the cytokine-induced gene signatures. While the approach has its limitations, since cytokine signatures were obtained from cultured keratinocytes, this result indicates that transcriptomic changes in psoriasis lesions are not merely extrinsic (reactive), but also intrinsic; this is in line with a recent study (11).
Psoriasis has a strong genetic component and GWAS have identified numerous genetic variations associated with psoriasis (3, 17, 24). We found that 107/2,365 DEGs identified in psoriatic keratinocytes were localized in the proximity of SNPs previously associated with psoriasis (3, 4, 17, 18), suggesting that genetic variations underlie part of the observed gene expression changes. A large proportion of these DEGs are localized on chromosome 1, where the epidermal differentiation complex (EDC) gene cluster, mapping to the psoriasis susceptibility locus PSORS4, is localized, comprising many genes of crucial importance for keratinocyte differentiation (25). Our findings confirm previously identified keratinocyte-related susceptibility genes, such as the LCE genes (25), and the NF-κB regulatory protein TNFAIP3. Notably, TNFAIP3 is an example for a psoriasis susceptibility gene, which was previously mostly associated to the immune system; however, its keratinocyte-specific deletion in mice leads to epidermal hyperproliferation and increased susceptibility to psoriasis-like skin inflammation (26). Our results showing its decreased expression in psoriatic keratinocytes supports the results obtained with the mouse model, and suggests that its decreased expression in keratinocytes can contribute to psoriasis. Our results also identify genes previously uncharacterized in psoriasis that overlap with psoriasis susceptibility regions and have altered expression in keratinocytes; some examples are GSDMA, which regulates epithelial homeostasis (27) and PPP1R15A, known to be downregulated in SCC and to regulate hyperproliferation/apoptosis (28). Our findings suggest that genetic variations affecting gene expression in psoriatic keratinocytes, in addition to those related to immune cell functions, contribute to psoriasis susceptibility.
Previous studies have shown alterations in gene expression occur in the non-lesional psoriasis skin, suggesting a “pre-psoriatic state”, possibly caused by genetic alterations (9). However, the contribution of keratinocytes to these changes has not been known. In our study, alterations in gene expression, even if mild, were detected already in non-lesional keratinocytes, suggesting that changes in non-lesional skin may partially be due to intrinsic, genetically determined, changes in the keratinocytes. These findings are in line with a recent study published (11) during the preparation of this manuscript, reporting alterations in the cultured keratinocytes derived from non-lesional psoriasis skin. The relatively subtle transcriptional changes in the non-lesional keratinocytes suggest that part of the genetic variations affect gene expression only upon certain stimuli, such as the inflammatory microenvironment present in psoriasis lesions.
Fifty-four transcription factors whose expression was altered in psoriatic keratinocytes and whose target genes were enriched among DEGs in psoriatic keratinocytes were identified. Among these were AP-1 (c-Jun, c-Fos, JunD, FosB), whose importance for psoriasis is evidenced by the psoriasis-like phenotype of keratinocyte-specific alterations of JunB/c-Jun (29), as well as several others (e.g. RORA, RORC, FOXO1, NF-κB p50 and p105, GATA3, E2F family, EGR) which have been implicated in psoriasis. It is noteworthy that several of the identified transcription factors (e.g. SOX4, KLF4, AP1, IRF4, STAT2/3) are located in previously characterized psoriasis susceptibility regions (4), thereby representing a mechanistic link between genetic susceptibility and molecular alterations in psoriatic keratinocytes. Consistent with our findings, AP-1 as well as KLF4 were identified as transcriptional regulators using cultured keratinocytes from psoriasis skin (11).
Importantly, several other transcription factors previously not characterized in the context of psoriasis, were identified in our analyses: e.g. TRPS1 (Transcriptional Repressor GATA Binding 1), HEY2 (Hes Related Family BHLH Transcription Factor With YRPW Motif 2) whose expression is induced by the Notch pathway (30), and PAX3 (Paired Box 3) involved in ectodermal differentiation pathways (31). The function of these transcription factors needs to be further investigated in the context of psoriasis as they may represent potential targets for topical antipsoriatic therapy.
In conclusion, this study identifies keratinocyte-specific transcriptomic changes in psoriasis lesional and non-lesional skin. These results complement previous studies describing transcriptomic changes in psoriasis skin, and indicate that keratinocytes are major contributors to molecular changes in psoriasis skin. Moreover, these results indicate that keratinocyte-specific alterations are in part intrinsic/genetically determined and in part extrinsic/reactive to the cytokine milieu.
subjects who took part in this study and those who contributed to the work, especially research nurse Helena Griehsel and Maria Lundqvist for assistance and careful handling of the samples and databases.
This work was funded by the Swedish Medical Research Council (Vetenskapsra?det), Swedish Psoriasis Association (Psoriasisfo?rbundet), Welander and Finsen Foundations /Hudfonden (Skin Foundation) and the Stockholm County Council (ALF). E.S. has received consultancy or speaker honoraria from Novartis, Abbvie, Eli Lilly, UCB and Pfizer Inc.


 Click to show fullsize
Click to show fullsize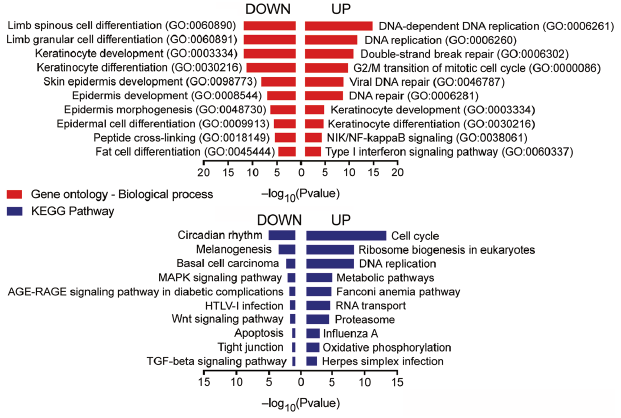 Click to show fullsize
Click to show fullsize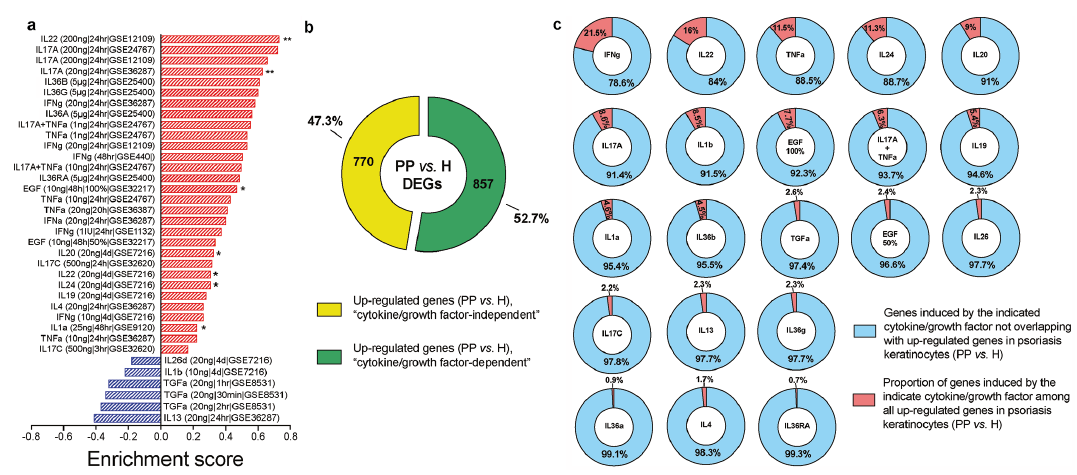 Click to show fullsize
Click to show fullsize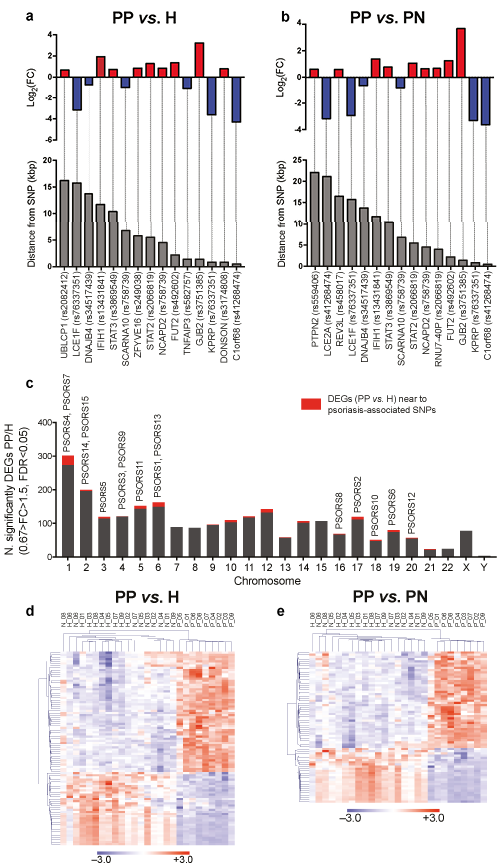 Click to show fullsize
Click to show fullsize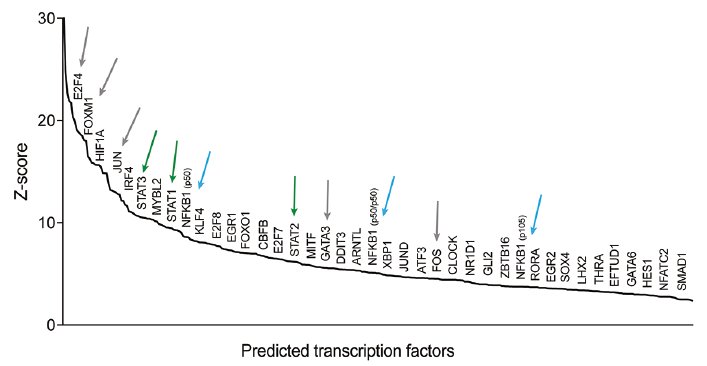 Click to show fullsize
Click to show fullsize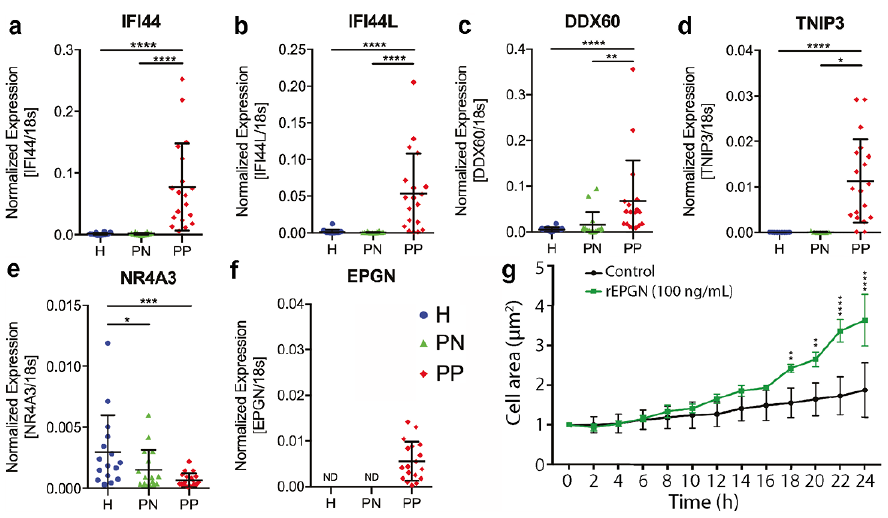 Click to show fullsize
Click to show fullsize