


Department of Dermatology, University of Kiel, Kiel, Germany
Staphylococcus epidermidis is an abundant skin commensal capable of activating cutaneous defense responses, such as induction of cytokines and antimicrobial peptides. To permanently colonize human skin and prevent inflammation S. epidermidis needs to control the induction of host defense mediators. We report here that S. epidermidis induces expression of the host regulator protein A20 in human keratinocytes, thereby controlling expression and release of interleukin-1 beta. siRNA-mediated knockdown of A20 expression strongly enhanced the induction of interleukin-1 beta gene expression and protein release in keratinocytes stimulated with S. epidermidis. Furthermore, siRNA-mediated knockdown of A20 resulted in enhanced gene expression and secretion of the antimicrobial peptide human beta-defensin-2 in keratinocytes facing S. epidermidis. Mechanistically, A20 negatively controlled S. epidermidis-induced activation of the transcription factor NF-kappaB. Together, these data indicate that S. epidermidis exploits A20 to attenuate cutaneous defense responses, which may help S. epidermidis to persist on human skin.
Key words: keratinocytes; A20 (TNFAIP3); interleukin-1 beta; beta-defensins; Staphylococcus epidermidis; cutaneous defense.
Accepted Oct 16, 2018; Epub ahead of print Oct 17, 2018
Acta Derm Venereol 2019; 99: XX–XX.
Corr: Maren Simanski, Department of Dermatology, University of Kiel, Rosalind-Franklin-Str. 7, DE-24105 Kiel, Germany. E-mail: msimanski@dermatology.uni-kiel.de
The abundant skin commensal Staphylococcus epidermidis induces innate defense mediators, such as IL-1 beta and the antimicrobial peptide hBD-2 in keratinocytes. This study shows that an exaggerated production of these defense mediators is controlled by the parallel S. epidermidis-mediated induction of the host molecule A20. A20 is a known inhibitor of inflammatory signalling, and dysregulation of A20 may be associated with chronic inflammatory diseases, such as psoriasis. Induction of A20 may help S. epidermidis to persist as a commensal on human skin and to avoid an increased inflammatory scenario. Targeted induction of A20 may be useful to treat specific cutaneous inflammatory conditions.
The Gram-positive bacterium Staphylococcus epidermidis is an abundant commensal of human skin and it is considered as an important member of the cutaneous microbiota (1, 2). Its presence on the skin is associated with risk of nosocomial S. epidermidis infections, especially infections associated with medical devices, such as catheter-related infections (3–5). However, under normal conditions S. epidermidis appears to contribute to cutaneous innate immunity. This is achieved by the production of antimicrobial factors, such as phenol-soluble modulins (PSMs), which are able to restrict the growth of potential pathogenic bacteria, such as Staphylococcus aureus (6). S. epidermidis is also able to secrete the serine protease Esp, which inhibits biofilm formation and destroys pre-existing biofilms of S. aureus (7). In addition, secreted products of S. epidermidis are able to induce the expression of antimicrobial peptides, such as human beta-defensin (hBD)-2 and -3 in keratinocytes (8–10). Induction of antimicrobial peptides in keratinocytes can also be achieved via IL-17A released by IL-17A+ CD8+ T cells that were previously primed by CD103+ dendritic cells upon sensing commensal bacteria, such as S. epidermidis (11).
The role of S. epidermidis for protective immunity has also been demonstrated in a germ-free mouse model. Germ-free mice were highly susceptible to intradermal infection with the parasite Leishmania major. When the skin of the germ-free mice was concomitantly colonized with S. epidermidis protective immunity against L. major was recovered. Immune responses triggered by the presence of S. epidermidis were mediated by IL-1 signalling, which promoted effector T-cell responses (12). Thus, IL-1 seems to be an important defense molecule produced in the skin upon contact with S. epidermidis.
To verify that keratinocytes facing S. epidermidis might
also represent a cellular source for the release of IL-1, in particular IL-1 beta, we stimulated human primary keratinocytes with living S. epidermidis. Stimulation of the keratinocytes with S. epidermidis induced the gene expression and release of IL-1 beta (see below). IL-1 beta is a powerful pro-inflammatory cytokine and its overexpression is associated with several inflammatory diseases (13, 14). Therefore we hypothesized that S. epidermidis-mediated induction of IL-1 beta in keratinocytes must be tightly regulated to avoid uncontrolled inflammatory scenarios induced by commensals, such as S. epidermidis in the absence of pathogens.
We report here that S. epidermidis induces the expression of A20 (“TNFalpha-induced protein 3”, TNFAIP3) in human primary keratinocytes, thereby controlling the expression and release of IL-1 beta. In addition, our data indicate that A20 also restricts the S. epidermidis-induced expression of the antimicrobial peptide human beta-defensin-2 (hBD-2). Thus, our study suggests that S. epidermidis exploits the host molecule A20 to dampen induction of host defense responses in keratinocytes facing S. epidermidis.
Human primary keratinocytes (Promocell, Heidelberg, Germany) were cultured in “Keratinocyte Growth Medium 2” (KGM2) plus supplements (Promocell) without antibiotics at 37 °C in a 5% CO2 atmosphere. The keratinocytes were used for stimulation at passage 3–5 and cultured in 12- or 24-well plates. Confluent keratinocytes were stimulated with S. epidermidis (identity verified by MALDI-TOF mass spectrometry; MALDI Biotyper, Bruker, Billerica, MA, USA) at a concentration of ~1×107 colony-forming units (CFU) per ml corresponding to a multiplicity of infection of ~10–20. Bacteria were centrifuged on cells at 350×g for 5 min, followed by incubation at 37°C for 3 h. Subsequently, the culture medium was removed and cells were washed twice with phosphate-buffered saline (PBS) to remove non-adherent bacteria. The cells were incubated for additional 17–19 h in culture medium supplemented with 200 μg/ml gentamicin. After stimulation, cell supernatant was harvested for determination of protein release by enzyme-linked immunoassay (ELISA) and cells were lysed for RNA isolation.
Primary human keratinocytes were transfected with 5 nM siRNA at a confluence of 40–60% using 1 µl transfection reagent HiPerFect (Qiagen, Hilden, Germany). After 16–20 h medium was removed and the keratinocytes were further cultured for 3 days until stimulation with S. epidermidis. Two different A20-specific siRNAs and appropriate non-silencing control siRNAs were used: “ON-TARGETplus SMARTpool” (L-009919-00 and Non-targeting Pool D-001810-10) and “SilencerSelect” (s14259 and non-silencing control siRNA 4390844) were purchased from Dharmacon (Lafayette, CO, USA) and Life Technologies (Carlsbad, CA, USA), respectively.
After stimulation, total RNA of the keratinocytes was isolated using the Crystal RNAmagic reagent according to the manufacturer’s protocol (Biolabproducts, Gödenstorf, Germany). 0.5 µg total RNA served as template for cDNA synthesis using an oligo dT primer and 12.5 units reverse transcriptase (PrimeScript RT Reagent Kit, TaKaRa Bio, Saint-Germain-en-Laye, France). The resulting cDNA served as template in a real-time PCR. Real-time PCR was performed in a StepOne Real Time PCR System (Applied Biosystem, Carlsbad, CA, USA) using SYBR Premix Ex Taq II (TaKaRa Bio, Saint-Germain-en-Laye, France) as previously described (15). The following intron-spanning primers were used: IL-1 beta: 5´- AAG CCC TTG CTG TAG TGG TG -3´ (forward primer) and 5´- GAA GCT GAT GGC CCT AAA CA -3´ (reverse primer); hBD-2: 5`-GCC TCT TCC AGG TGT TTT TG-3` (forward primer) and 5`-GAG ACC ACA GGT GCC AAT TT-3` (reverse primer). All quantifications were normalized to the house keeping gene RPL38 (ribosomal protein L38) using the primer pair: 5`-TCA AGG ACT TCC TGC TCA CA -3` (forward primer) and 5`-AAA GGT ATC TGC TGC ATC GAA -3` (reverse primer). Standard curves were generated for each primer set with serial dilutions of cDNA. Relative expression is shown as a ratio between expression of the specific gene and RPL38 gene expression.
Secretion of IL-1 beta levels in the cell culture supernatants were determined by an IL-1 beta enzyme-linked immunoassay (ELISA) (R&D Systems; Minneapolis, MN, USA) according to the manufacturer’s protocol. The detection limit of the IL-1 beta ELISA was 3.4–7.8 pg/ml. Analysis of hBD-2 concentration was determined by a hBD-2 ELISA with antibodies purchased from Peprotech, as previously described (16). The detection limit of the hBD-2 ELISA was 0.6 ng/ml.
Analysis of NF-κB was performed using a NF-kappaB firefly luciferase reporter plasmid, as described previously (15). Luciferase activity was calculated by the amount of firefly luciferase activity normalized to the amount of renilla luciferase activity.
For analysing proteolytic processing of pro-IL-1 beta keratinocytes were transfected with a luciferase-based inflammasome and protease activity reporter plasmid termed iGLuc (17). This reporter, which contains Gaussia luciferase fused to pro-IL-1 beta, secretes luciferase activity in the supernatant of the keratinocytes upon proteolytic cleavage of pro-IL-1 beta. Keratinocytes were transfected with this reporter plasmid (300 ng/well) 24 h before stimulation using 1 µl of the transfection reagent Fugene HD (Promega).
GraphPad Prism 6.07 was used for statistical analysis (unpaired 2-tailed Student’s t-test or 1-way analysis of variance (ANOVA), followed by Holm-Sidak’s multiple comparisons test).
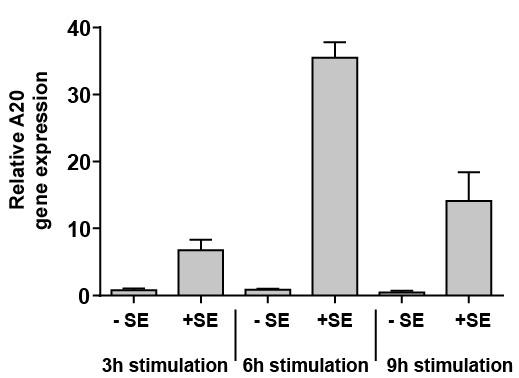
Stimulation of human keratinocytes with S. epidermidis revealed a rapid induction of A20 gene expression, as measured by real-time PCR (Fig. 1). Time kinetics with measurements after 3, 6 and 9 h showed increased gene expression after 3 h.
Fig. 1. Staphylococcus epidermidis induces gene expression of A20 in human primary keratinocytes. Human primary keratinocytes were stimulated for the indicated time with S. epidermidis (+SE) or left unstimulated (–SE). Gene expression of A20 was analysed by real-time PCR. Bars are means ± standard error of the mean (SEM) of 3 stimulations.
To investigate whether A20 might have a regulatory function on the S. epidermidis-mediated induction of IL-1 beta, primary keratinocytes were transfected with A20-specific siRNA to downregulate A20 expression. This resulted in 68% knockdown of A20 gene expression as revealed by real-time PCR (Fig. 2a). Induction of IL-1 beta gene expression by S. epidermidis was significantly increased in the keratinocytes treated with A20-siRNA (Fig. 2b). Similarly, S. epidermidis-induced IL-1 beta protein release was also enhanced in the A20-knockdown keratinocytes compared with the cells treated with a control siRNA (Fig. 2c). To verify these results we used a second A20-specific siRNA, which resulted in a 71% downregulation of A20 gene expression (Fig. 2d). In concordance with the other results the keratinocytes transfected with this second A20-siRNA responded with an enhanced S. epidermidis-induced IL-1 beta gene expression and protein release (Fig. 2e, f).
Fig. 2. Staphylococcus epidermidis-mediated induction of IL-1 beta in human keratinocytes is controlled by A20. (a–c) Human primary keratinocytes were transfected with a control siRNA or an A20-specific siRNA (Dharmacon) and stimulated with S. epidermidis (+SE) or left unstimulated (–SE). Knockdown efficiency (KD) of A20 gene expression (a) and IL-1 beta gene expression (b) were analysed by real-time PCR. Secretion of IL-1 beta was determined by ELISA (c). (d–f) Human primary keratinocytes were also transfected with another A20-specific siRNA (Life Technologies) and again stimulated with S. epidermidis. Knockdown efficiency (KD) of A20 gene expression (d) and IL-1 beta gene expression (e) were analysed by real-time PCR. (f) Secretion of IL-1 beta was determined by enzyme-linked immunoassay (ELISA). Bars are means ± standard error of the mean (SEM) of 6 stimulations (***p < 0.001, **p < 0.01, *p < 0.05, n.s.: not significant; 1-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test).
We also investigated whether A20 might influence inflammasome activity in the keratinocytes stimulated with S. epidermidis. To this end we used the luciferase-based inflammasome reporter iGLuc, which detects cleavage of pro-IL-1 beta, resulting in increased luciferase activity (17). Using keratinocytes transfected with this reporter we observed that S. epidermidis induced release of luciferase activity, indicating proteolytic cleavage of pro-IL-1 beta (Fig. S1). However, this was not significantly influenced by gene knockdown of A20, indicating that A20 has no direct effect on the processing of pro-IL-1 beta in the S. epidermidis-stimulated keratinocytes (Fig. S1). Thus, our data indicate that, in human keratinocytes, A20 negatively regulates expression of IL-1 beta on the transcriptional level, but does not influence proteolytic release of IL-1 beta.
This study aimed to determine whether NF-κB is affected by A20 in the keratinocytes stimulated with S. epidermidis. To this end the keratinocytes were transfected with a NF-κB luciferase reporter plasmid to monitor NF-κB activation. Treatment of the human primary keratinocytes with S. epidermidis induced NF-κB activation as measured by an increase in luciferase activity (Fig. 3b). This S. epidermidis-mediated NF-κB activation was significantly increased when A20 gene expression was downregulated by siRNA (Fig. 3a, b). In addition, we measured alterations in gene expression of the NF-κB inhibitor IkBalpha (NFKBIA) because it has been demonstrated that activation of NF-κB correlated with gene induction of NFKBIA. Therefore, gene induction of NFKBIA can be used as a marker of NF-κB activation (18). In line with the NF-κB gene reporter luciferase results, S. epidermidis induced NFKBIA in keratinocytes confirming activation of NF-κB by S. epidermidis (Fig. 3d). Again, NF-κB activation by S. epidermidis was increased in keratinocytes with siRNA-mediated downregulation of A20 gene expression (Fig. 3c, d). Together these results illustrate that A20 restricts S. epidermidis-induced NF-κB activation.
Fig. 3. A20 controls Staphylococcus epidermidis-induced NF-κB activation. (a, b) Human primary keratinocytes were transfected with a control or an A20-specific siRNA. 1 day before stimulation cells were additionally transfected with a NF-κB firefly luciferase reporter plasmid and a renilla luciferase control plasmid. Cells were then treated with S. epidermidis (+SE) or left untreated (–SE). (a) Knockdown efficiency (KD) of A20 gene expression was determined by real-time PCR. (b) NF-κB activity was determined by analysis of luciferase activity which was calculated as the ratio between firefly and renilla luciferase activities in each sample. (c and d) Human primary keratinocytes were transfected with a control or an A20-specific siRNA and stimulated with S. epidermidis (+ SE) or left untreated (–SE). (c) Knockdown efficiency (KD) of A20 gene expression was determined by real-time PCR. (d) Gene expression of NFKBIA was measured by real-time PCR. Bars are means ± standard error of the mean (SEM) of 3 (a and b) or 6 (c and d) stimulations (***p < 0.001, **p < 0.01, *p < 0.05; 1-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test).
Because NF-κB regulates induction of the defense molecule hBD-2 we hypothesized that S. epidermidis may also induce hBD-2 in an A20-controlled manner. In line with this hypothesis hBD-2 gene induction in keratinocytes stimulated with S. epidermidis was strongly increased upon siRNA-mediated downregulation of A20 gene expression (Fig. 4a, b). Thus, hBD-2 protein release was also significantly enhanced in keratinocytes treated with A20-siRNA as compared with cells treated with control siRNA (Fig. 4c). Knockdown of A20 gene expression with another A20-specific siRNA (Fig. 4d) also resulted in increased S. epidermidis-induced hBD-2 gene expression (Fig. 4e) and protein secretion (Fig. 4f). These results identify hBD-2 as another A20-controlled factor.
Fig. 4. Staphylococcus epidermidis-mediated induction of hBD-2 in human keratinocytes is controlled by A20. (a–c) Human primary keratinocytes were transfected with a control siRNA or an A20-specific siRNA (Dharmacon) and stimulated with S. epidermidis (+SE) or left unstimulated (–SE). Knockdown efficiency (KD) of A20 gene expression (a) and hBD-2 gene expression (b) were analysed by real-time PCR. Secretion of hBD-2 was determined by enzyme-linked immunoassay (ELISA) (c). (d–f) Human primary keratinocytes were also transfected with another A20-specific siRNA (Life Technologies) and again stimulated with S. epidermidis. Knockdown efficiency (KD) of A20 gene expression (d) and hBD-2 gene expression (e) were analysed by real-time PCR. Secretion of hBD-2 was determined by ELISA (f). Bars are means ± standard error of the mean (SEM) of 3 stimulations (***p < 0.001, **p < 0.01, *p < 0.05, n.s.: not significant; 1-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test).
Knockdown of A20 expression by siRNA increased basal NF-κB activity in unstimulated keratinocytes, and this was further induced by S. epidermidis stimulation (Fig. S2a,b). Knockdown of A20 also slightly induced basal hBD-2 gene expression, which was further induced by stimulation with S. epidermidis (Fig. S2d). Basal hBD-2 peptide release, as well as basal IL-1 beta gene and protein expression, were not significantly affected by A20 knockdown (Fig. S2c,d). Knockdown of A20 led to a significant increased induction of IL-1 beta and hBD-2 expression upon S. epidermidis treatment (Fig. S2c,d).
The gene TNFAIP3 encodes A20, an important signal molecule that negatively regulates NF-κB activation via its deubiquitinase activity, thereby limiting excessive inflammatory processes (19). The importance of A20 as a master regulator of inflammation is reflected by various studies describing a link between A20 dysregulation and the onset of autoimmune and inflammatory diseases, such as type 1 diabetes, systemic lupus erythematosus, inflammatory bowel disease, ankylosing arthritis, Sjögren’s syndrome and rheumatoid arthritis (20, 21). Although the role of A20 in innate immunity is still emerging, there is evidence that A20 interferes with innate immune signalling. In particular, it has been reported that A20 is able to interfere negatively with Toll-like receptor activation in macrophages and with nucleotide-binding oligomerization domain containing 2 (NOD2) responses in mice (22, 23). In contrast, the role of A20 in cutaneous defense is not well defined. It has been reported that gene expression of A20 is induced upon wounding (24). An induction of cytokines in keratinocytes treated with poly (I:C) was inhibited by A20 overexpression (25). Furthermore, epidermal A20 is implicated in the proper control of normal epidermal appendage development (26). However, the function of A20 in epidermal innate defense is not well understood.
This study analysed the role of A20 in human keratinocytes stimulated with S. epidermidis. S. epidermidis induced A20 within 3 h, indicating that S. epidermidis induces A20-mRNA in a direct manner. Furthermore, RNA interference experiments with 2 different specific A20-siRNAs showed that induction of IL-1 beta gene expression by S. epidermidis, as well as the protein expression of IL-1 beta, was attenuated by A20. This is in concordance with a recent study reporting that A20-deficient mouse macrophages showed an increased gene expression of pro-IL-1 beta, which was associated with an increased release of IL-1 beta. The same study also reported increased gene expression of NLRP3 in A20-deficient mouse macrophages thereby catalysing activation of the NLRP3 inflammasome (27). Of note, the results of the current study demonstrate that the proteolytic processing of IL-1 beta in the S. epidermidis-stimulated keratinocytes was not influenced by A20. Thus, our data indicate that, in human keratinocytes, A20 dampens the S. epidermidis-mediated IL-1 beta gene induction and protein release, but does not influence proteolytic processing of IL-1 beta.
It has been shown that transcription factor NF-κB is crucial for gene induction of IL-1 beta by various stimuli, including phorbol ester and lipopolysaccharide (28). There is also evidence that S. epidermidis is able to activate NF-κB in human keratinocytes (29). On the other hand, A20 is able to negatively regulate NF-κB-mediated gene expression and to downregulate NF-κB signalling (30, 31). Therefore, we analysed whether NF-κB is affected by A20 in the keratinocytes stimulated with S. epidermidis. Our results illustrate that A20 restricts S. epidermidis-induced NF-κB activation. These data suggest that induction of A20 by S. epidermidis reduces S. epidermidis-mediated NF-κB activation, thereby controlling excessive activation of NF-κB. This may represent a novel mechanism for how a commensal on the skin surface restricts inflammatory scenarios induced by the commensal itself.
Human beta-defensin-2 (hBD-2) is another NF-κB-regulated innate defense molecule, and induction of hBD-2 by bacteria can be mediated by NF-κB (16, 32). As discussed above, S. epidermidis is able to activate NF-κB in keratinocytes, and this activation was negatively regulated by A20. Therefore we hypothesized that S. epidermidis may also induce hBD-2 in an A20-controlled manner. Indeed, our results revealed that S. epidermidis dampens exaggerated induction of hBD-2 through parallel induction of A20.
It is intriguing to speculate that other bacteria also engage A20 to regulate induction of cytokines and antimicrobial peptides in keratinocytes. Indeed, our unpublished preliminary data suggest that A20 is implicated in the regulation of hBD-2 induction in keratinocytes stimulated with the Gram-negative bacterium Pseudomonas aeruginosa. This observation suggests that it is a general principle of several bacteria to induce A20 to downregulate innate defense, a hypothesis that remains to be verified in future studies.
A20 has recently gained novel interest in the context of allergy. It has been reported that farm dust and endotoxin stimulate A20 induction, thereby protecting the airways from house dust mite-induced asthma (33). The same study also showed that levels of A20 were lower in airway epithelia derived from patients with asthma compared with healthy controls. In addition, a single-nucleotide polymorphism (SNP) in the A20 gene was associated with an increased risk of asthma and eczema (33). The association with eczema strengthens the hypothesis that A20 may play an important regulatory role in skin barrier homeostasis.
In gut enterocytes LPS-mediated induction of A20 blocked responses to re-stimulation with bacterial products, such as LPS and CpG DNA (34). Reduction of the microbiota in the gut by antibiotics decreased A20 levels (34). Moreover, it is known that A20-deficient mice develop severe intestinal inflammation early in life (35, 36). These data indicate an important role for A20 in the development of tolerance in the intestinal epithelium to avoid commensal-induced inflammation (34). Our data make it likely that such a mechanism may also exist in the skin; however, this remains to be shown in future studies.
Taken together, our data suggest that the co-induction of A20 by S. epidermidis controls an exaggerated S. epidermidis-mediated production of innate defense mediators, such as IL-1 beta and hBD-2. Thus, through exploitation of a host regulator protein S. epidermidis attenuates cutaneous innate defense responses, which may help S. epidermidis to persist as a commensal on human skin. Future studies should evaluate whether the targeted induction of A20 may be useful to treat specific cutaneous inflammatory conditions.
The authors thank Heilwig Hinrichs and Cornelia Wilgus for technical assistance. The S. epidermidis clinical isolate 12034044 was kindly provided by Professor R. Podschun (Department of Infection Medicine, Kiel, Germany). The authors thank Professor Dr Veit Hornung for providing the iGLuc plasmid. This study was supported by a grant from the German Research Foundation given to Maren Simanski (SI 2015/2-1).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize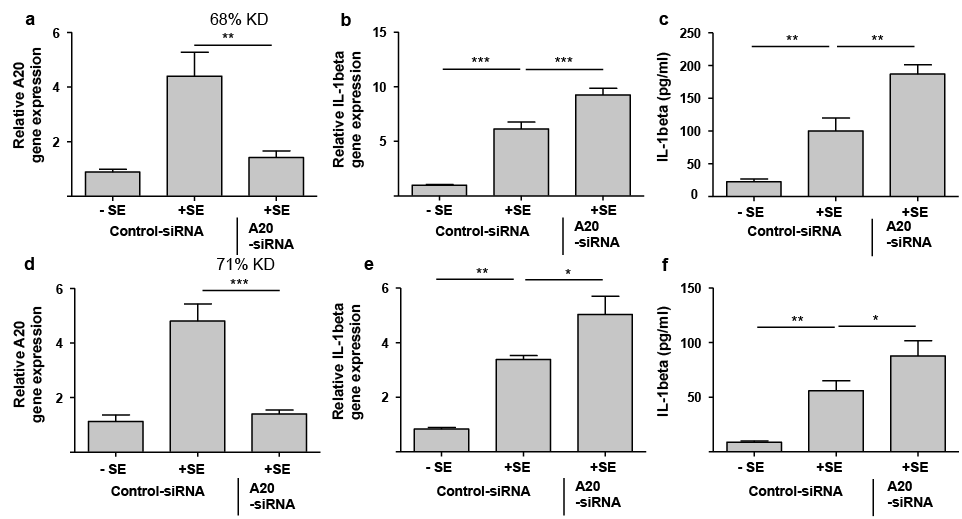 Click to show fullsize
Click to show fullsize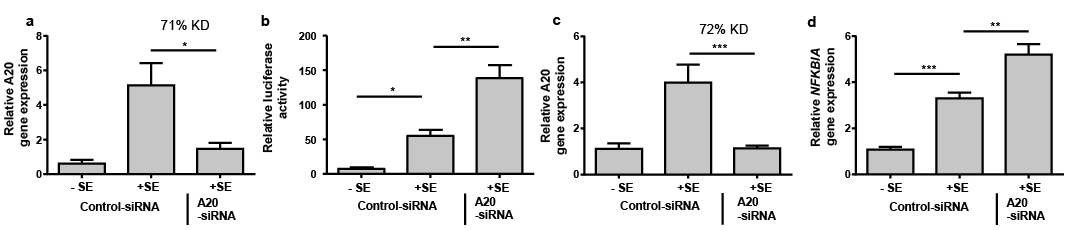 Click to show fullsize
Click to show fullsize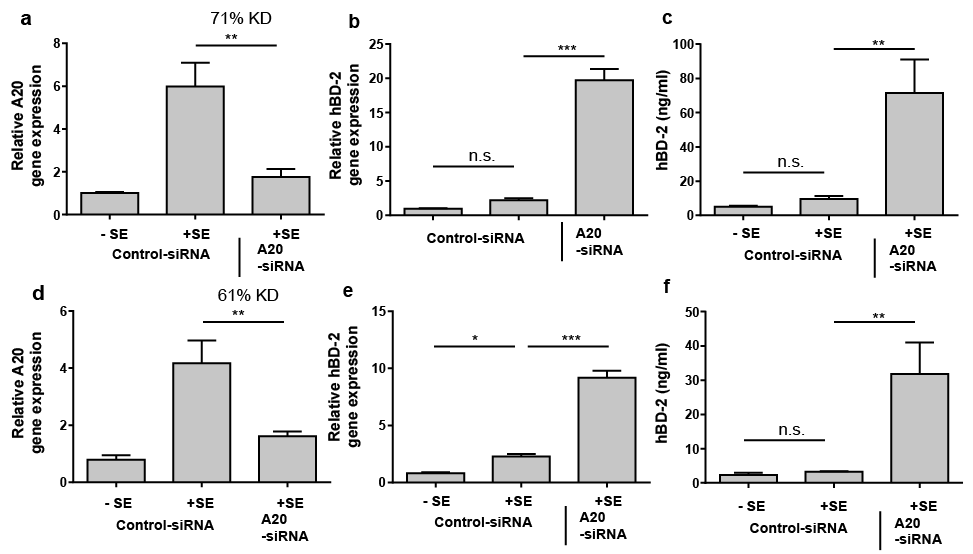 Click to show fullsize
Click to show fullsize