


1Unit of Anatomic Pathology, Department of Molecular Medicine, IRCCS San Matteo Foundation, University of Pavia, Pavia, 2Unit of Dermatology, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Via Pace 9, Milan, 3Department of Oncology and Hematology, and 4Department of Pathophysiology and Organ Transplantation, University of Milan, Milan, Italy. E-mail: jessica.gliozzo@gmail.com
Accepted Jan 7, 2019; E-published Jan 9, 2019
Xanthoma disseminatum (XD) is an extremely rare cutaneous, normolipidemic, proliferative disorder of the Mononuclear Phagocyte System. It was first described by Von Gräfe and Virchow but recognized as a distinct entity by Montgomery & Osterberg in 1938 (1). Approximately 140 cases are reported in the English literature. Clinically, XD is characterized by a symmetrical skin eruption of reddish papules or nodules, with predilection for the head, neck, flexural and intertriginous areas. These lesions, initially isolated, tend to slowly merge into large yellowish plaques, during years. The mean age at presentation is 30 years and male cases outnumber female ones by a ratio of 2:1. XD is generally thought to be a benign disorder with a chronic progressive course (2–4). Caputo et al. (5) recognized 3 clinical variants of XD: 1) a self-healing form; 2) a persistent form and 3) a progressive multi-system (MS) form. The report rate of this entity is becoming even lower now than in past years, perhaps due to its high diagnostic overlap with Erdheim-Chester Disease (ECD) (6, 7).
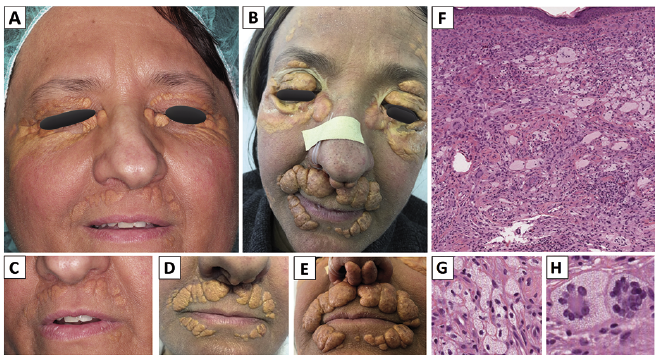
We hereby describe the case of a previously healthy 47-year-old Caucasian woman presented to our attention in January 2011, with a 2-year long history of cephalic xanthomas. Her lesions consisted of yellow periocular and perioral plaques extending to the zygomatic area as well as small yellow hard-elastic nodules growing next to the inner canthi (Fig. 1). Complete radiological, hematological (blood count, metabolic, renal and liver functions/profiles) and endocrinological examinations all tested negative and failed to reveal any systemic involvement. During the following 7 years, the lesions displayed a slow but progressive extension with symmetrical involvement of the whole face (Fig. 1). She never developed diabetes insipidus or any other signs of systemic involvement. Previous therapies (hydroxychloroquine and cyclo-phosphamide) and surgeries (including CO2 laser therapy) led to constant recurrences and subsequent regrowth of lesions, together with surgery-related keloid scars, resulting in high mechanical, psychological and sociological impairment. Skin biopsy (Fig. 1) showed a polymorphic dense upper- and mid-dermal infiltration consisting of large histiocytes, admixed with few lymphocytes and neutrophils. Histiocytes were mostly characterized by large nuclei often with prominent nucleoli and an abundant pale cytoplasm. Foamy, Touton and Langhans cells were also present, focally in large numbers. There were no signs of epidermotropism. Immunohistochemical profile of the histiocytes was positive for CD4, CD11c, CD14, CD68/PGM1, CD163, fXIIIa, vimentin, lysozyme, fascin but tested negative for CD1a, CD34 and CD207/langerin. Ki-67 stained 1–5 % of the cells. BRAFV600E analysis failed to reveal any alteration. Sixteen years after the first manifestation, the patient is alive and in a good clinical condition, although with chronic persistent evolution of the disfiguring lesions.
Informed consent was obtained in accordance with local ethical guidelines and with the Helsinki Declaration of 1975.
Fig. 1. Clinical and histopatho-logical features of the patient. The patient came to our attention with yellowish plaques limited to periocular and perioral regions (A, C). During the first 2 years of follow-up the lesions slowly increased in volume and extended to columella (D) and after 5 years they presented as thick tumor/nodular lesions and confluent plaques extending to the forehead, zygoma, eyelids, outer nostrils and mentolabial sulci (B, E). Skin biopsy revealed a dense and polymorphic histiocytic infiltrate (F) made up of large cells with abundant and often foamy cytoplasm (G) and multinucleated giant cells of the Touton (H) and Langhans type, admixed with small lymphocytes and neutrophils.
Histiocytoses have recently been reclassified in 5 different groups based on a combination of clinical, radiographic, histopathological and molecular findings. XD belongs to the “xanthogranuloma-family” of the C(utaneous)-group, due to its prevalent skin involvement and the polymorphic histiocytic infiltrate made of foamy cells and giant multinucleated cells (6–8).
Clinical presentation of XD is highly heterogeneous as location and evolution of the skin lesions may be highly variable. Mucous membrane manifestations appear in half cases, as well as diabetes insipidus. The lesions may cause severe mechanical impairment (e.g. mechanical blindness, respiratory obstruction or difficulty in defecation). Involvement of other organs (i.e. bones, kidneys, lymph nodes, nervous system) is described, although less frequent (2–4).
Differential diagnosis of XD is highly problematic, same as for other non-Langerhans-cell histiocytoses (NLCHs) (see Table SI). Purely cutaneous (single-system) XD cases generally involve multiple areas including eyelids, neck, axillae, elbows, groin and perianal region and should be clinically differentiated from adult xanthogranuloma, based on the number and diffusion of the lesions. Differential diagnosis may be more challenging for multi-system-XD, as many clinical-pathological features coincide with the recently defined diagnostic criteria for ECD. Those two histiocytoses are part of the same spectrum of entities, if not synonyms, as a matter of fact (9, 10). Our patient displays two very uncommon features for XD, namely the face-limited localization and the tumor-like lesions. The isolated cephalic involvement has been previously reported in only 3 cases (11). The limitation to head and neck region is seen in other NLCH but it is the predominant feature of benign cephalic histiocytosis (BCH), a pediatric histiocytosis presenting with a self-limiting papular eruption. XD could be differentiated from BCH on the basis of epidemiological, clinical and histopathological findings. One hypothesis could link such circumscribed localization to some regional (dis-)immune factors (10).
The development of nodular/tumor-like lesions, is otherwise seen in progressive nodular histiocytosis (PNH), another rare C-histiocytosis characterized by a florid symmetrical proliferation of hundreds of large nodules/tumors all over the cutaneous surface. XD and PNH are indistinguishable from a histopathological point of view, although PNH may also display a spindle-shaped histiocyte morphology (12).
The hyper-activation of the MAPK-pathway seems to play an important role in histiocytoses and has been proposed as diagnostic markers for some entities (e.g. ECD) (13). To the best of our knowledge, the present report is the only one that characterizes XD on a molecular level. Even though the differential benefit of molecular analysis in histiocytoses has recently been denied, molecular characterization is important in order to gather information about the etiology and possibly prognostic stratification in such a rare disorder (13).
There is no standard treatment for XD, although, several therapeutic strategies have been described (14). One review described a subset of cases with spontaneous resolution of lesions (15). Whereas several other papers reported progression and/or death of patients even after aggressive chemotherapy. Consequently, the best therapeutic approach still needs to be weighed against the patient’s clinical severity (4, 11).
In conclusion, our patient’s features conform to the clinical heterogeneity of histiocytoses and show how differential diagnosis, prognostic stratification and therapeutic approach may be incredibly troublesome. Future works should point out the overlaps between NLCH and revise their nomenclature, allowing for an easier approach to therapeutic decisions.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize