


Departments of 1Dermatology and 3Human Genetics and Molecular Biology, Nagoya University Graduate School of Medicine, Nagoya, 466-8550, 2Division of Dermatology, Japanese Red Cross Nagoya Daiichi Hospital, and 4Department of Genetics, Research Institute of Environmental Medicine, Nagoya University, Nagoya, Japan. E-mail: miro@med.nagoya-u.ac.jp
Accepted Jan 16, 2019; E-published Jan 17, 2019
Galli–Galli disease (GGD) is a rare autosomal dominant genodermatosis exhibiting reticulated hyperpigmentation and scaling erythematous papules mainly on the great skin folds (1). GGD is considered to be an acan-tholytic variant of Dowling–Degos disease (DDD) (2). Mutations in KRT5 (3) and POGLUT1 (4) have been reported in GGD patients. In contrast, DDD is a rare autosomal dominant genetic pigmentary disorder characterized by dot-like or reticulate, slightly depressed, sharply demarcated brown macules particularly affecting the flexures and other major skin folds. KRT5 was identified as a causative gene of DDD in 2006 (5), and recently POFUT1 (6) and POGLUT1 (4) were clarified as additional causative genes. Finally, PSENEN was identified as a causative gene of DDD with hidradenitis suppurativa (7).
Here, we report a Japanese female GGD case with a previously unreported POGLUT1 mutation. As far as we know, the present case is the first non-Caucasian GGD patient with a POGLUT1 mutation.
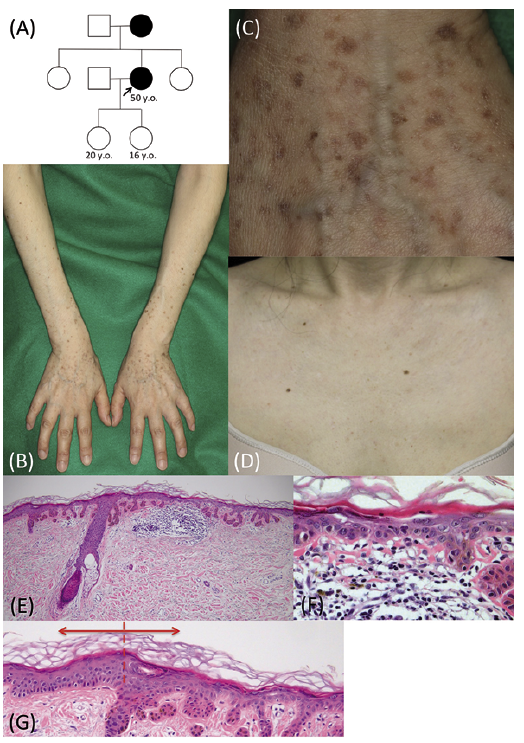
A 50-year-old Japanese woman had noticed small pigmented spots on the extremities in her late twenties that had increased in number gradually. According to her, her mother had similar pigmented macules. Neither of her daughters (ages 20 years and 16 years) had similar pigmented macules. She visited our neighbor hospital and was referred to our department for diagnosis of the hyperpigmented macules mainly on the extremities (Fig. 1A).
She showed irregularly shaped, slightly keratotic, sharply demarcated grayish-brown macules of 2 to 5 mm in diameter on the extremities and the trunk. The lesions were more sparse on the trunk than on the extremities. Neither a reticulate arrangement of the macules nor depression of the macules was observed in the present patient. The macules on the extremities were mainly seen on the dorsal side. The number of macules on the extremities gradually increased from the proximal to the distal regions. The flexor sides of the knees and elbows and other major skin folds were spared (Fig. 1B). A few erythematous lesions with itching were intermingled with the brown macules. Palmoplantar pits and pigmentation were absent. She showed fewer macules on the face and the V-neck site, and the neck was completely spared (Fig. 1D). Faint depigmented macules were also seen on the extremities and at the V-neck site.
A skin biopsy obtained from a pigmented macule on the forearm showed elongation of the rete ridges with diffuse pigmentation in the epidermis and hyperkeratosis without parakeratosis. Under high magnification, acantholysis and atypism of keratinocytes were seen in the epidermis. The lesional epidermis was thinner than that of the surrounding normal skin (Fig. 1G). Patchy inflammatory cell infiltration and pigmentary incontinence were seen in the upper dermis (Fig. 1E, F). From the above information, the differential diagnoses were GGD, which is an acantholytic variant of DDD, and xeroderma pigmentosum (XP), variant type. We confirmed our diagnosis by genetic analysis.
The Ethics Committee of the Nagoya University Graduate School of Medicine approved the studies described below. All studies were conducted according to the principles of the Declaration of Helsinki. The participant gave written informed consent.
Whole-exome sequencing was performed with genomic DNA from the patient’s peripheral blood sample. We found a heterozygous frameshift mutation, c.1013delTinsAAC, p.Ile338Lysfs*27, in POGLUT1, which was confirmed by Sanger sequencing (Fig. 2A). We made the final diagnosis of GGD due to the POGLUT1 mutation (OMIM: #615696).
Fig. 1. The clinical and histopathological features of the patient. (A) The pedigree of the proband’s family. (B-F) Skin manifestations of the proband. The macules on the extremities are mainly on the dorsal side. The number of macules on the extremities gradually increases from the proximal to the distal regions (B). Irregularly shaped, slightly keratotic, sharply demarcated grayish-brown macules of 2 to 5 mm in diameter are seen on the dorsal hands (C). Only a small number of macules are seen at the V-neck site. Faint depigmented macules are seen at the V-neck site (D). (E–G) Histopathologically, elongation of the rete ridges with diffuse pigmentation is observed in the epidermis and patchy inflammatory cell infiltration is seen in the superficial dermis (E). Under high magnification, acantholysis and atypism of epidermal keratinocytes are seen and pigmentary incontinence is observed in the upper dermis (F). The lesional epidermis (right side) is thinner than that in the adjacent normal skin (left side), and hyperkeratosis without parakeratosis is seen in the stratum corneum (dotted line, a border of the lesion) (G).
Fig. 2. Causative mutation analysis in POGLUT1 of the present patient with Galli–Galli disease (GGD). (A) The mutation c.1013delTinsAAC, p.Ile338Lysfs*27 (in exon 10) detected by whole-exome sequencing was confirmed by Sanger sequencing analysis of POGLUT1 in the patient’s gDNA from peripheral blood leukocytes. (B) A schematic of the structure of human protein O-glucosyltransferase 1 enzyme encoded by POGLUT1. Protein O-glucosyltransferase 1 contains a signal peptide (SP), a CAP10 domain, and a Lys-Asp-Glu-Leu (KDEL)-like endoplasmic reticulum retention signal (KTEL). The present mutation is thought to abolish the C-terminal in 13 % of the CAP10 domain and to abolish the entire KTEL domain.
Generally, RAK is included in differential diagnoses of GGD/DDD. In the present case, we clinically excluded RAK and DDD due to POFUT1 mutations because the patient had none of the characteristic features of RAK (8–10) or DDD due to POFUT1 mutations (6): depressed reticular pigmentation on the dorsal hands and feet, palmoplantar pits, and intertriginous reticulate brown macules. In addition, her disease had a late onset, unlike the prepubertal onset of RAK.
To date, 16 unrelated cases of GGD/DDD due to POGLUT1 mutations have been reported (4, 11, 12). All have been Caucasian, and the present case is the first non-Caucasian patient with a POGLUT1 mutation to be reported in the literature, as far as we know. Unlike the previously reported cases, our case showed grayish dark brown macules, which were similar to seborrheic keratosis and senile freckle.
Both POGLUT1 and POFUT1, which encode protein O-glucosyltransferase 1 enzyme and protein O-fucosyltransferase 1, respectively, are known to modify NOTCH proteins. However, unlike the patients with POFUT1 mutations, the patients with GGD/DDD due to POGLUT1 and KRT5 mutations have widespread skin lesions, but the flexor areas of the extremities are spared (4, 12). In addition, they show acantholysis in the epidermis (4, 12). Thus, the aberrant modification of NOTCH proteins might not be a pathogenic mechanism that is shared by patients with DDD due to POGLUT1 and POFUT1 mutations. The mutant protein O-glucosyltransferase 1 derived from the POGLUT1 mutant allele in the present case has 87% of the CAP10 domain, and the present patient showed skin lesions almost exclusively in the extremities (Fig. 2B). In contrast, the mutant protein O-glucosyltransferase 1 derived from the POGLUT1 mutant alleles with p.Arg218* and p.Arg272* in the two cases in the literature (11, 12) has 39% and 60% of the CAP10 domain, respectively, and both patients had extensive lesions on the trunk and the extremities. These findings might suggest genotype/phenotype correlations in POGLUT1 mutations.
Non-Caucasian patients with GGD/DDD due to POGLUT1 mutations might have phenotypes that differ from those of Caucasian patients, because Caucasian and non-Caucasian patients differ in skin color. In this context, the further accumulation of non-Caucasian GGD/DDD patients with POGLUT1 mutations is thought to be required.
This study was funded in part by JSPS KAKENHI Grant Number 17K10240 to MK, by a Japan Agency for Medical Research and Development (AMED) JP17ek0109281 to TO and by a grant from the Kao Melanin Workshop to MK.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize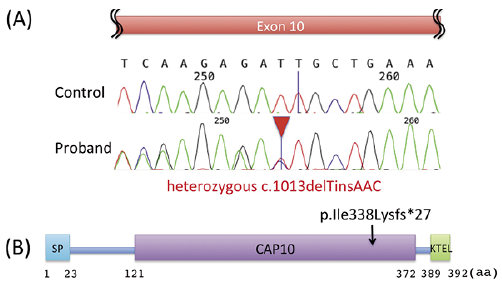 Click to show fullsize
Click to show fullsize