


1Center for Chronic Pruritus, Department of Dermatology, University Hospital Münster, Münster, 2Fachklinik Bad Bentheim, Department of Dermatology, Bad Bentheim, 3Institute of Biostatistics and Clinical Research, University of Münster, Münster, 4Department of Dermatology and Allergy, Charité –Universitätsmedizin Berlin, 5Clinical Research Center, Department of Dermatology, University Medical Center, Mainz, 6Hannover Medical University, Department of Dermatology, Allergology, and Venereology, Hannover, 7Clinics of Dermatology, Allergy Faculty of Medicine and Health Science, University of Oldenburg, Klinikum Oldenburg AöR, Oldenburg, 8Department of Dermatology, Jena University Hospital, and Department of Dermatology, SRH Wald-Klinikum Gera, Jena, and 9Centre for Clinical Trials, Medical Faculty of the University of Münster, Münster, Germany
#Both authors contributed equally to this paper.
The aim of this multicentre, randomized, double-blind, placebo-controlled, cross-over, phase-II study was to determine the antipruritic effect of aprepitant vs. placebo in 58 patients with anti-histamine-refractory chronic pruritus in chronic nodular prurigo. Patients were randomized to receive either first oral aprepitant 80 mg/day or placebo for 4 weeks. Following a 2-week wash-out phase, the patients were crossed-over to receive the other treatment for 4 weeks. Primary efficacy criterion was the intra-individual difference between mean itch intensity (visual analogue scale) at baseline compared with the end of treatment period. Prurigo lesions, pruritus course, quality of life, patient benefits, and safety were secondary parameters. No significant differences were found between aprepitant treatment and placebo for any of the parameters investigated. Under the experimental conditions of the study, aprepitant, 80 mg daily for 4 weeks, did not have an antipruritic effect in patients with chronic prurigo. (DRKS00005594; EudraCT Number: 2013-001601-85).
Key words: pruritus; itch; chronic nodular prurigo; substance P; neurokinin receptor 1; NK1 antagonist.
Accepted Jan 16, 2019; E-published Jan 17, 2019
Acta Derm Venereol 2019; 99: XX–XX.
Corr: Sonja Ständer, Center for Chronic Pruritus, Department of Dermatology, University Hospital Münster, Von-Esmarch-Str. 58, DE-48149 Münster, Germany. E-mail: sonja.staender@uni-muenster.de
Chronic nodular prurigo is characterized by multiple highly pruritic cutaneous nodules and a high impact on quality of life. The molecule substance P appears to play a significant role in the pathway of chronic pruritus. This randomized, double-blind, placebo-controlled, cross-over trial aimed to find out if using the drug aprepitant to block the substance P pathway could influence chronic pruritus in chronic nodular prurigo. However, the results showed no significant difference between aprepitant and placebo.
Chronic pruritus (CP) (of over 6 weeks’ duration) has a lifetime prevalence of up to 25.5% (1) and considerably impairs quality of life; thus it represents a worldwide burden (2, 3). Although the medical care of patients with CP has improved over recent years, through establishment of specialized itch clinics, a classification system, and well-defined treatment guidelines (4, 5), the currently available treatment modalities are not sufficiently efficacious in many patients with CP (6, 7). In addition, many of the recommended therapies exhibit adverse effects and cannot be used on a long-term basis. Therefore, there is high level of need for new treatment options that target the biological mechanisms of CP.
Substance P (SP), which binds to neurokinin receptor 1 (NKR1), is a major mediator of pruritus (8, 9). NKR1 is expressed both in the central nervous system and in the skin (10). Animal models have demonstrated an anti-pruritic effect by SP inhibition at the NKR1 (11, 12). In an NC/Nga mouse model, oral treatment with the NKR1-antagonist aprepitant, reduced the level of serum immunoglobulin E (IgE), tissue SP levels, and cutaneous infiltration of regulatory T cells (13). The clinical relevance of NKR1-antagonism in humans has been shown in several case series of acute and CP of various origin, using the inhibitor aprepitant (14–21). In an open-label, not placebo-controlled proof-of-concept study, 20 patients with therapy-refractory CP of various origins experienced significant (p < 0.001) antipruritic effect within one week of monotherapy with aprepitant 80 mg once daily (22). This also included patients with chronic nodular prurigo (CNPG), who showed good responses (22). However, there is a lack of evidence regarding the antipruritic effect of aprepitant from controlled studies. The aim of this study was to close this gap, and to compare the effect of aprepitant with placebo, not only regarding symptom relief, but also considering pruriginous lesions, quality of life and patient benefits, as recommended by the International Forum for the Study of Itch (IFSI) (23).
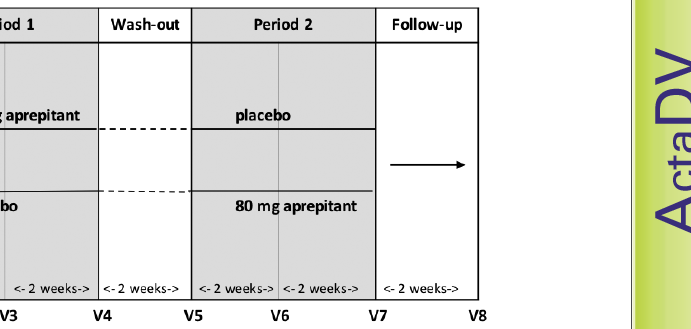
This investigator-initiated, prospective, multicentre, randomized (1:1), double-blind, placebo-controlled, cross-over, phase-II clinical study was conducted in 5 dermatological hospitals in Germany. After giving informed consent, the patients were randomized to receive orally either 80 mg aprepitant (Emend®, Merck Sharp & Dohme Corp., Kenilwoth, New Jersey, USA) once daily or placebo for 4 weeks (Period 1). Following a 2-week wash-out phase (26–37 times longer than the half-life), patients crossed over, receiving the other treatment for 4 weeks (Period 2). Finally, the patients were transferred into a 2-week follow-up phase. Overall, the patients were invited to attend 8 visits (for details see Fig. 1).
The study was approved by the ethics committees at the central coordinating centre (Münster) and at each of the participating trial sites. The trial was registered at the German Clinical Trials Register (registration number DRKS00005594). The study was performed in accordance with the Declaration of Helsinki and the requirements of the guidelines for good clinical practice (GCP).
Fig. 1. Study design. Following screening and baseline there was the first treatment period for 4 weeks (Period 1). After a 2-week wash-out phase, patients crossed over, receiving the alternative treatment in the second treatment period for 4 weeks (Period 2). At the end there was a follow-up after 2 weeks.
The eligibility of adult patients (18–70 years of age) with CNPG of dermatological, systemic or mixed origin with a mean baseline pruritus visual analogue scale (VAS 0–10) of ≥ 7 during one of the two previous days was assessed. Patients were required to have therapy-refractory pruritus (no reduction of > 2 VAS points) with more than 4 weeks’ systemic antihistamine therapy. Women of child-bearing potential had to agree to adequate birth control methods during the study.
Exclusion criteria were: patients who had CP of paraneoplastic, neurogenic, psychogenic origin and those with additional severe skin inflammation necessitating anti-inflammatory therapy (e.g. urticaria, bullous pemphigoid, acute generalized flaring up of atopic dermatitis), or only localized pruritus; patients who had other unstable or uncontrolled significant medical conditions, infections, current malignant disease, use of therapies that could influence the study outcome for up to 2 weeks (topical corticoids, systemic antihistamines) or 4 weeks (systemic corticosteroids, immunomodulators, opioid receptor agonists/antagonists, antidepressants, immunosuppressants, CYP3A4 inducers, antimycotics, topical calcineurin inhibitors, antibiotics, antiseptics, or phototherapy) before onset of the study, had an allergy to any of the treatment’s ingredients, participated in another investigational clinical study up to 4 weeks before baseline, or were pregnant or lactating.
Patients were randomized into 2 arms in a 1:1 ratio, to receive first aprepitant and then placebo, or vice versa. Randomization was performed by a blinded biometrician who was not otherwise involved in the study. The randomization schedule was stratified block-wise by trial site with concealed block size. Allocation concealment was provided by sealed envelopes. The randomization process was monitored and reviewed during the entire enrolment phase. To ensure blinding, the aprepitant and placebo capsules were overcoated at the pharmacy of the University Hospital of Mainz with identical size 00 gelatine capsules.
The primary efficacy (PE) end-point was the intra-individual difference in mean itch intensity (VAS) in the last 24 h before and after each treatment period. According to the 2×2 cross-over design, each patient was supposed to provide the PE for both treatment periods 1 (PE1) and 2 (PE2) (Fig. 1). Thus, negative PE1 and PE2 values would indicate improvement in the respective periods 1 and 2. Furthermore, in case of PE1–PE2<0, the improvement would be greater in period 1 and vice versa.
Secondary efficacy variables included: baseline adjusted, i.e. intra-individual difference of visit 4 minus baseline visit 2, and of visit 7 minus “baseline” visit 5 (visit-obtained according to patient global assessment (PGA) (24)) worst itch (VAS) (25); visit-obtained, baseline adjusted itch, burning, and stinging, as evaluated on a 5-point verbal rating scale (VRS) (25); global and dynamic score according to PGA at visits 4 and 7, respectively; time course changes in VAS, VRS and global/dynamic score according to PGA (visit-obtained); itch time course changes in VRS and in numeric rating scale (NRS (26); mean and worst itch) and a global question concerning the presence of pruritus (PGA) (diary-obtained); time course changes in quality of life assessed with the Dermatological Quality of Life Index (DLQI) (27), ItchyQoL (28, 29) and the Hospital Anxiety and Depression Scale (HADS) (26) (visit-obtained); patient benefit index as assessed by the patients at the visits (PBI-P; 0–4 scale) (30); Prurigo Activity Score items (PAS) (31) (predominance of prurigo type, complete skin healing, number of prurigo lesions, activity and percentage of healed lesions) and the total PAS (visit obtained); and adverse events (AEs). Itch, grading of itch, mean, and worst, were all assessed daily by the patients in the electronic diary (ItchApp© (32)) starting at visit 2 and continuing until visit 8. The secondary efficacy analysis evaluated the intra-individual differences under aprepitant in comparison with placebo. Furthermore, a subanalysis of response in atopic and non-atopic patients was conducted, as well as a comparison of the results obtained by PAS and VAS with those from VRS and NRS in order to analyse the sensitivity of the tools and to validate the diary-obtained measurements.
The safety period under aprepitant was defined as the days from visit 2 to visit 5 for all patients who received aprepitant in period 1, and the days from visit 5 to visit 8 for all patients who received aprepitant in period 2. The safety period under placebo was defined analogously. The safety period before baseline included all days before visit 2. For safety analyses, every AE (with seriousness/without seriousness; with/without possible correlation with study medication) were calculated for each safety period and in total. Corresponding analyses were also conducted for adverse reactions (ARs), serious adverse events (SAEs), and non-SAEs. The outcomes were discussed with the Data and Safety Monitoring Board.
Based on the treatment difference from a previous study with similar inclusion criteria (22), and assuming that 10% of patients would be non-evaluable (e.g. drop-outs), a target sample size of 26 patients per arm (n = 52 in total) was calculated to provide 90% power to detect a treatment effect with a 2-sided 5% level of significance.
The intention-to-treat (ITT) set (or full analysis set) included all randomized patients and the per-protocol (PP) set all patients without major protocol violations. The latter were identified by the coordinating investigator before unblinding. The safety set consisted of all patients who received at least one dose of study medication (aprepitant or placebo).
All statistical analyses were predefined in the statistical analysis plan before unblinding and carried out accordance with ICH guideline E9 using SAS software, version 9.4 for Windows (SAS Institute, Cary, NC, USA). The primary efficacy analysis was performed according to the ITT principle to obtain confirmatory evidence. The 2 arms were compared regarding the CROS outcome (CROS(PE)=PE1–PE2) by a stratified Wilcoxon–Mann–Whitney U test on a 2-sided 5% significance level with strata defined by the patients’ atopic/non-atopic predisposition. For both arms, the median and interquartile range of the PE1 and PE2 values were computed. For each arm, boxplots of the CROS(PE) values were drawn, and the PE1 values were plotted against the PE2 values (scatterplots). For sensitivity analysis, the same analyses were performed using the PP set.
All secondary efficacy analyses were performed according to the ITT and the PP principle. Descriptive analyses were carried out using common measures of location and scale. All statistical tests (2-sided) were intended to deliver exploratory results and conducted in due consideration of the cross-over design, if appropriate. Regarding metric endpoints, the treatments were therefore compared according to the CROS principle described above. Comparisons regarding categorical end-points were carried out using the Mainland-Gart test. Moreover, the treatments were also compared regarding the primary and secondary efficacy endpoints from period 1 only. Such comparisons were performed according to a simple parallel group design, using common quantile-based descriptive and non-parametric inductive methods.
Safety analysis was conducted for all patients who received at least one dose of study medication, using exploratory statistical methods.
Between June 2014 and January 2016 (Fig. 2), a total of 67 patients with CNPG were screened. Of these, 58 met the eligibility criteria (ITT) and were randomized either first to aprepitant (arm A, n = 30, 12 females, mean age 57.3 ± 10.7 years, mean VAS mean 72.5 ± 19.5) or first to placebo (arm B, n = 28, 15 females, mean age 53.2 ± 13.1 years, mean VAS mean 76.2 ± 17.0) (Table I). In arm A, 2 patients were lost to follow-up and 9 had major protocol deviations. In arm B, 24 patients received the allocated intervention and 5 of them had major protocol deviations. Thus, the PP population consisted of 19 patients per arm (Fig. 2). Since all 30 patients in arm A and 24 of the patients in arm B received at least 1 dose of aprepitant, the safety set consisted of 54 patients. Baseline demographic and clinical characteristics and treatment compliance were similar between the 2 arms (Table I).
Fig. 2. Flow diagram showing the progress of all patients through the trial. After screening 67 patients with CNPG 58 met the eligibility criteria. In the end the per protocol population consisted of 19 patients per arm.
Table I. Demographic and clinical characteristics at baseline
Primary efficacy analysis could not confirm a significant difference between the 2 arms as to the CROS(PE) values, either in ITT (p = 0.7) or in PP (p = 0.8) analysis (Table II; Fig. 3). Subgroup analyses conducted in patients with atopic and non-atopic CNPG did not reveal either significant differences between aprepitant and placebo (Table II).
Table II. Primary and secondary efficacy results (intention-to treat; ITT and per-protocol; PP) including subgroup analyses of atopic and non–atopic patients
Fig. 3. Course of mean itch intensity (visual analogue scale (VAS) in the last 24 h, primary endpoint) by study arm (line: 50 quartile; dashed line: 25 and 75 quartiles).
All secondary efficacy analyses, which were also conducted on both the ITT and the PP set, delivered similar to the primary efficacy analysis results. Analysis of the VAS and VRS endpoints and of the DLQI and ItchQoL scores did not reveal any differences between aprepitant and placebo. However, in both arms, patients reported an improvement in the first period (visit 2–4), which did not reduce significantly during the wash-out phase, but also did not progress further in the second period (Fig. 3).
No differences between active treatment and placebo were seen for HADS, PBI-P (Table II), PAS items (Table III) and for global and dynamic scores. Patients used rescue medication either in both periods or never, with no difference between the arms.
Analysis of the electronic diary (ItchApp©)-obtained endpoints also did not show any difference between aprepitant and placebo. The vast majority of patients reported itch every day regardless of being on treatment with aprepitant or placebo. Differences regarding the reported itch grading could also not be observed. Some patients mentioned improvement during the first few days, but there was no difference between active- and placebo-treated patients. Further exploratory analysis of PE1, responder rates, and of the frequency of patients having at least 75% healed pruriginous lesions also did not show differences between aprepitant and placebo (Table III).
Table III. Development of Prurigo Activity Score (PAS) items (ITT) during the study
Safety analysis did not indicate any differences between the 2 arms of this study. The total number of AEs was 32 under aprepitant treatment and 38 under placebo, while 3 AEs occurred before baseline. Most AEs were mild to moderate and only one, under placebo, was severe (severe back pain). No serious adverse event (SAE) occurred under aprepitant. Two SAEs occurred under placebo; both in one patient, and neither were severe (hospitalization twice due to tonsillectomy). Ten non-severe ARs were observed under aprepitant and 7 under placebo. No serious AR occurred.
In the past, the antipruritic effect of NK1 antagonism with aprepitant was demonstrated in mouse models (11, 12) and in several case series in patients with acute and chronic pruritus (14–21). The objective of this study was to confirm the antipruritic and symptom-alleviating effect of aprepitant 80 mg daily vs. placebo in patients with severe CP in CNPG. A clinical benefit of aprepitant was assumed in patients with CNPG with respect to encouraging case reports (22) and a known increased density of SP-positive skin nerve fibres and high serum levels of SP have both been found in patients with CNPG (33).
However, this study failed to confirm the hypothesis, as there was no difference between the aprepitant and placebo arms in terms of reduction in pruritus, improvement in pruriginous lesions or quality of life. Also, the post-hoc subanalyses regarding the onset of antipruritic action in the first days, or response in the atopic subgroup did not show differences between aprepitant and placebo for any of the parameters evaluated. These results were unexpected, especially considering the 4-week treatment period and the fact that the dosage of 80 mg/day has been shown to optimally block NKR-1 (34).
In the first treatment period, a reduction in pruritus intensity was observed in both groups (aprepitant: 20% reduction, with a reduction of 1.5 VAS points; placebo 14.5% reduction, with a reduction of 1.1 VAS points). However, in the second treatment period the intensity increased by 3.8% under aprepitant or showed no changes in the placebo group. Based on a recent meta-analysis of clinical trials of patients with dermatological pruritic conditions (atopic dermatitis, urticaria and psoriasis) a placebo effect of 24% reduction in pruritus intensity from baseline could be expected (35). In addition, a VAS or NRS reduction of at least 3 points is considered a meaningful change in patients with CP (36). According to the crossover design of this study, patients knew that they would, in any case, receive the drug and a placebo. As a consequence (negative or positive) expectations and nocebo effects might have hindered the generation of an antipruritic effect in the current study, as observed under real-world conditions in open-label use of aprepitant. Our patients had a long duration of CNPG, had previous ineffective therapies, and were exposed for the first time to a study procedure. Also, the burden of patients with CNPG is higher than in other skin diseases (37) as they have more comorbidities with higher impact on quality of life (38). Taken together, these facts could explain why there is a high level of negative expectation and a lack of trust in novel therapies. This could explain that, in the first treatment period (visit 2–4), a slight decrease was observed in both arms, which disappeared completely in the second period.
Another explanation for the failure to confirm the hypothesis could be an insufficient sample size estimate. After withdrawal of consent or protocol deviations 19 patients per group could be analysed. This number could be too small to detect a 4-point decrease in the VAS, which is a sign of response.
However, during the 4-week treatment with the active drug, aprepitant appeared safe. Safety analyses of (serious) adverse events and reactions did not indicate any differences between aprepitant and placebo. Aprepitant has been tested and was also well tolerated in clinical trials with over 2,000 patients, in doses ranging from 40 to 240 mg per day for durations of up to 4 weeks (39–43).
Similar results have been published recently for the use of aprepitant in patients with atopic dermatitis in an open randomized controlled trial (RCT) (44) and in patients with Sézary syndrome in an RCT with cross-over design (45). In 19 patients with atopic dermatitis, there was no improvement in the extent of the disease, intensity of pruritus or scratching movements found when aprepitant, 80 mg daily for 7 days, was added to topical therapy with a moderately strong steroid and a moisturizer (44). Also, no antipruritic effect was found in 5 patients with Sézary syndrome receiving aprepitant 80 mg daily for 7 days (45).
Recently published data from a phase-II RCT has shown a significant dose-dependent reduction in intensity of pruritus after a 6-week treatment with the NKR-1 antagonist serlopitant, which can be used for a prolonged interval in patients with CP and CNPG (46). A phase-III trial may verify these promising results.
In conclusion, the current study, the first to evaluate the antipruritic effect of aprepitant under controlled conditions, could not confirm the efficacy of aprepitant vs. placebo. The hypothesis of aprepitant and NKR1 antagonism as an antipruritic therapy could not be validated for a 4-week treatment period in patients with CNPG.
The authors gratefully acknowledge funding from the German Federal Ministry of Education and Research (BMBF – Bundesministerium für Bildung und Forschung; grant number: 01KG1305). We thank Sonja Baier, Stefanie Dickmänken, Dr Trude Butterfaß-Bahloul from the Centre for Clinical Trials Münster for their support in conducting the study and Helena Karajiannis for her assistance in the preparation of the manuscript.
We also thank Prof Dr Matthias Augustin, Dr Andreas Kremer, Prof Dr Thomas Mettang, Dr Antje Jahn and Prof Dr Stefan Schneider for participating in the Data Safety and Monitoring Board.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize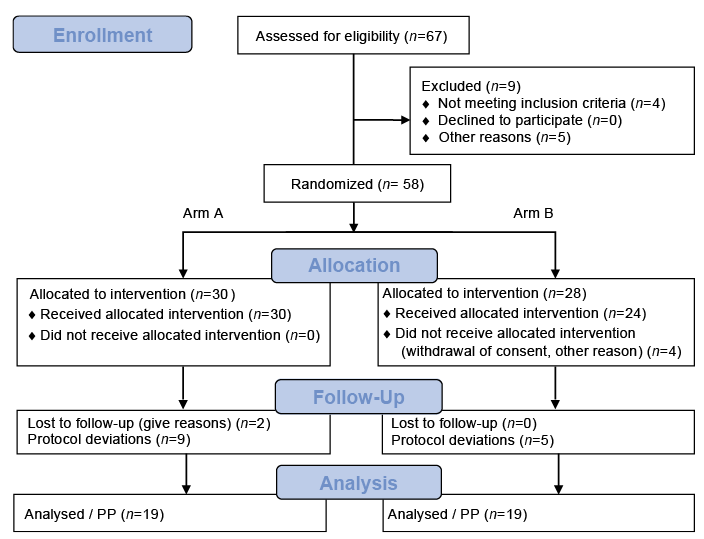 Click to show fullsize
Click to show fullsize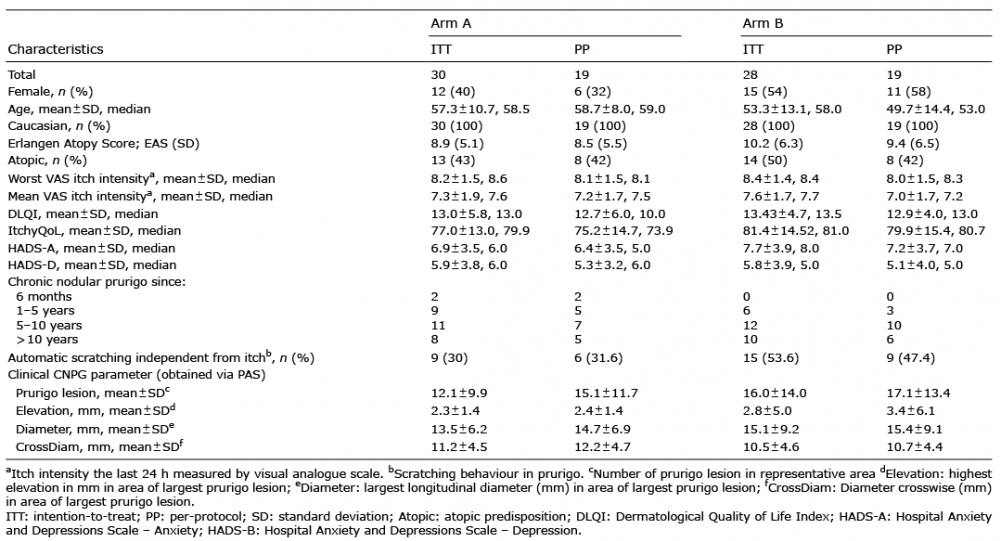 Click to show fullsize
Click to show fullsize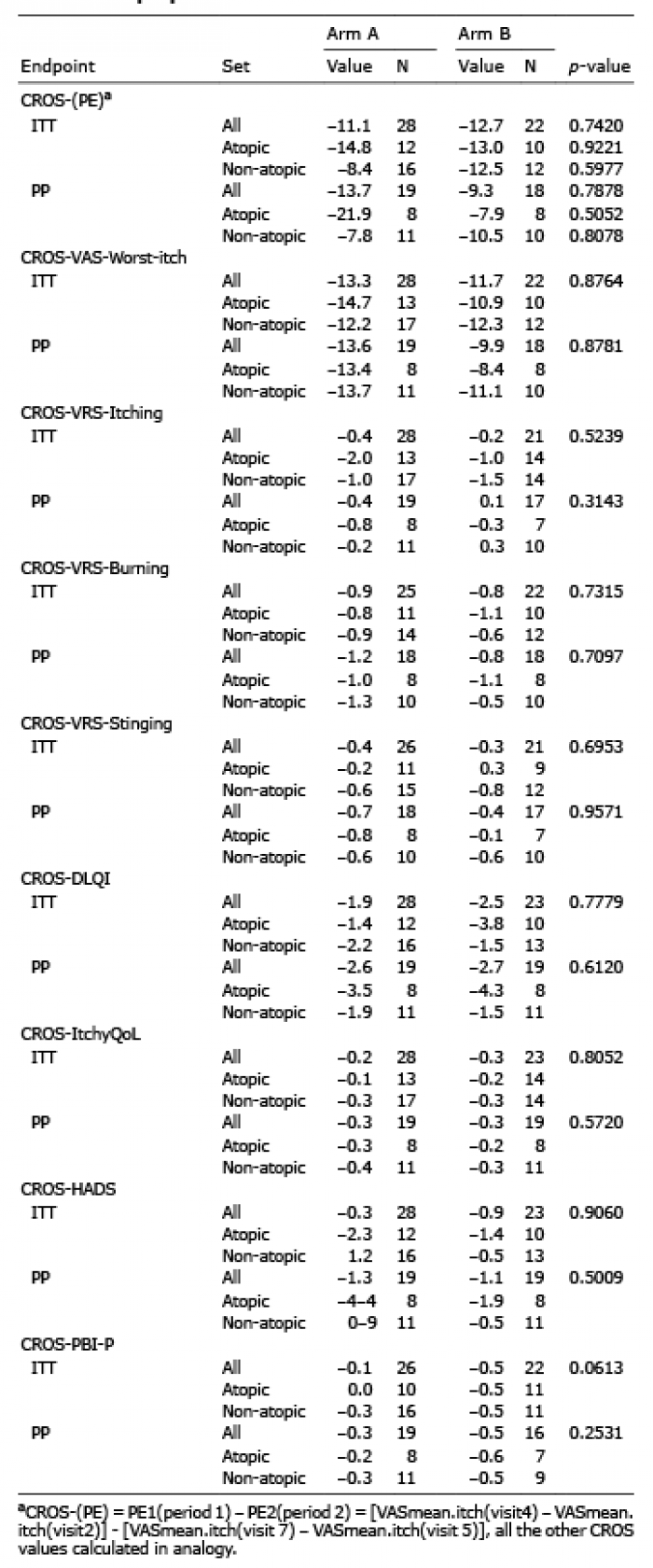 Click to show fullsize
Click to show fullsize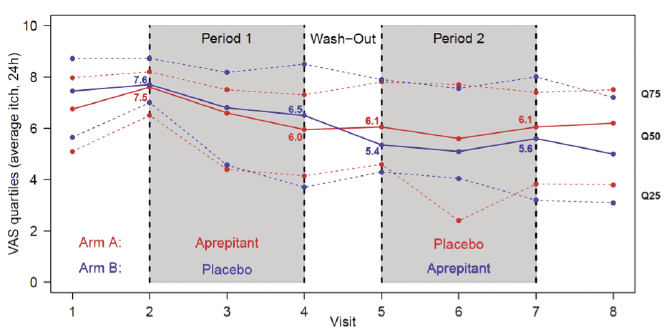 Click to show fullsize
Click to show fullsize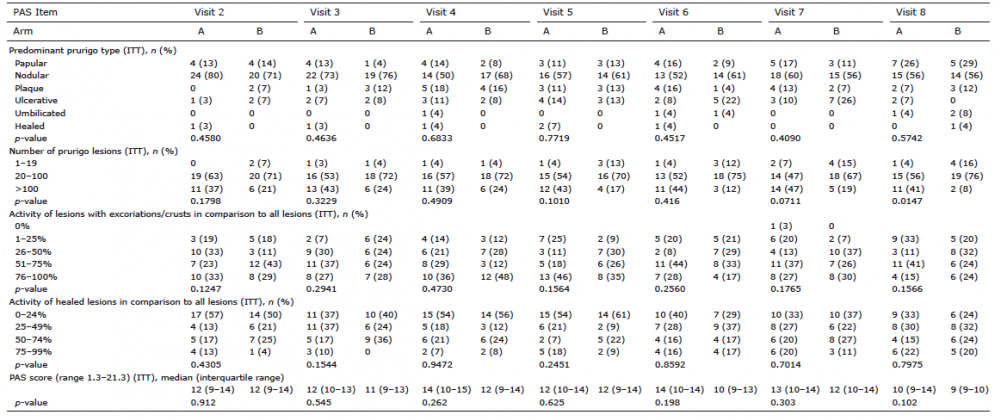 Click to show fullsize
Click to show fullsize