


Department of Dermatology and Allergy Centre, Odense University Hospital, Odense, Denmark
Skin cancer has become the most common type of cancer worldwide as a result of environmental exposure and medical treatments. A small group of patients are genetically predisposed to skin cancer and this article is intended as a diagnostic tool when encountering patients with multiple skin cancer lesions. The disorders are described with clinical characteristics, genetics and management. The most common syndromes associated with basal cell carcinoma are: Gorlin–Goltz syndrome, Rombo syndrome, and Bazex-Dupré-Christol syndrome. Multiple squamous cell carcinomas can be related to: xeroderma pigmentosum, Ferguson-Smith, Muir-Torre syndrome, Mibelli-type porokeratosis, keratitis-ichthyosis-deafness syndrome, Rothmund-Thomson syndrome, Bloom syndrome, and epidermodysplasia verruciformis. Malignant melanoma can be inherited, as in familial atypical multiple mole melanoma syndrome.
Key words: genodermatoses; skin cancer; basal cell carcinoma; squamous cell carcinoma; hereditary skin cancer.
Accepted Jan 16, 2019; E-published Jan 17, 2019
Acta Derm Venereol 2019; 99: XX–XX
Corr: Juliane Schierbeck, Department of Dermatology and Allergy Centre, Odense University Hospital, Vesterbro 116, 1th, DK-5000 Odense C, Denmark. E-mail: J.schierbeck@gmail.com
This article reviews hereditary skin syndromes that cause an increased risk of skin cancer development. It is important for physicians treating skin cancer to be aware of hereditary causes, especially when examining patients with multiple cancerous lesions with no obvious explanation. This article describes clinical features, genetic descriptions and management suggestions for hereditary syndromes associated with skin cancer, and includes clinical images from our practice.
Skin cancer is the most common type of cancer worldwide, with more than 15,000 patients annually in Denmark (which has a population of ~5.8 million) (1). Skin cancer is often caused by environmental exposure to ultraviolet radiation (UVR), immunosuppressive therapy or radiotherapy.
A small, and often overlooked, group of patients are genetically predisposed to develop skin cancer, sometimes associated with internal malignancies. These hereditary skin conditions, or genodermatoses, are often clustered, with multiple family members showing symptoms, al-though de novo mutations are also not uncommon. Awareness of these disorders is therefore essential for early diagnosis and treatment, as well as for identification of potentially affected family members. Early identification and diagnosis is crucial to the outcome and prognosis. Skin symptoms are easier to recognize, whereas visceral malignancies are more difficult and slower to identify. The first healthcare practitioners to see and treat these patients are general practitioners and dermatologists, who have a responsibility in recognizing and facilitating further genetic investigations and primary care, as described below.
This literature review focuses on hereditary causes of basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and malignant melanoma (MM). The review will serve as a tool in diagnosing and treating patients with multiple skin cancers.
Gorlin–Goltz syndrome (GGS), also known as Gorlin syndrome, naevoid basal cell carcinoma syndrome or multiple naevoid basal cell epithelioma, jaw cysts and bifid rib syndrome, is an autosomal dominant condition causing unusual facial appearances (mandibular prognathia, lateral displacement of the inner canthus, frontal and biparietal bossing), dental cysts, palmar pits and a predisposition for BCC. Other cardinal features are calcification of the falx cerebri, medulloblastoma, kyphoscoliosis, rib anomalies, cleft lip/palate, eye anomalies, milia and syndactyly (2). Two major and 1 minor criteria or 1 major and 3 minor criteria are necessary to determine the diagnosis, as shown in Table I.
Table I. Diagnostic criteria for Gorlin–Goltz syndrome
Binkley & Johnson were the first to suggest a correlation between dental cysts, partial agenesis of the corpus callosum, a bifid rib, an ovarian fibroma and epithelioma adenoides cysticum in 1951 (3). It was, however, the oral pathologist and human geneticist Robert J. Gorlin and the dermatologist Robert Goltz, who, in 1960, published and described the specific syndrome, which consists of multiple naevoid basal cell epitheliomas, jaw cysts and bifid ribs (4).
Molecular genetics and pathophysiology. Recent studies in molecular genetics have proven GGS to be caused by mutations in the PTCH1 gene on chromosome 9q22, the PTCH2 gene on chromosome 1p32, or the SUFU gene on chromosome 10q24-q25, encoding for proteins in the hedgehog signalling pathway, controlling growth and tissue development. The condition is autosomal dominant with complete penetrance, although approximately 10% of patients with GGS do not develop BCCs (5). The prevalence is reported to be 1 in 56,000–164,000 people, with an even sex distribution.
Characteristics. Patients with GGS often start developing BCCs between puberty and the age of 35 years, but other cutaneous characteristics, such as palmar pits, should also raise suspicion. The primary diagnostic criteria are odontogenic keratocysts, palmoplantar pits, calcification of the falx cerebri, medulloblastoma, a first-degree relative with GGS and multiple BCCs (2). Minor criteria include rib anomalies, other specific skeletal malformations including macrocephaly, cleft lip and/or palate, ovarian/cardiac fibroma, lymphomesenteric cysts and ocular abnormalities. Palmar pits in childhood are considered a very strong indicator, along with skeletal abnormalities, such as bifid ribs, frontal bossing and hypertelorism. Patients with GGS may develop from a few BCCs to several hundreds of BCCs during their lifetime (Fig. 1 A, B).
Fig. 1. (A, B) Patient with Gorlin–Goltz syndrome and multiple basal cell carcinomas (BCC) of the scalp and neck. This patient has had more than 600 tumours removed. (C) Hypotrichosis as seen in Rombo syndrome. (D) Milia on the cheek, as seen in Bazex-Dupré-Christol syndrome.
Michaelsson, Olsson & Westermark were the first to describe Rombo syndrome (RS), in 1981, when they described a family with vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, BCCs and peripheral vasodilation with cyanosis (6). The condition is extremely rare and only a few cases have been reported since then.
Molecular genetics and pathophysiology. The genetic mutation in RS has yet to be determined, but male-to-male transmission has been described in a family with transmission through 4 generations, suggesting autosomal dominant inheritance (7).
Characteristics. The syndrome presents in childhood with a reticular pattern of skin atrophy on the cheeks, preauricular area and forehead, along with cyanotic reddening of the skin. In adulthood whitish-yellowish, milia-like papules develop, along with telangiectatic vessels. Defective or completely missing eyelashes and eyebrows are also seen in adulthood. BCCs develop in the 3rd or 4th decade of life and are a consistent complication throughout life (8) (Fig. 1 C).
In 1964 Bazex, Dupré & Christol first described the condition Bazex-Dupré-Christol syndrome (BDCS) as an X-linked dominant syndrome affecting hair follicles and skin, along with an increased risk of developing BCCs. The condition is very rare; only approximately 20 families have been reported (9).
Molecular genetics and pathophysiology. A recent study has revealed that BDCS might be caused by mutations in the ARCT1 gene, resulting in an aberrant activation of the Hedgehog signalling pathway (10). BCCs have been described in the first decade of life, although they most commonly present in the second decade onwards. The female to male ratio is 2:1 (11).
Characteristics. Patients with BDCS are generally diagnosed based on a combination of hypotrichosis, multiple milia primarily on the face, follicular atrophoderma and multiple BCCs. Hypohidrosis and facial hyperpigmentation are also described as early-onset manifestations. The follicular atrophoderma is located mainly on the dorsa of the hands, giving a characteristic orange-peel appearance. BCCs are reported as early as the age of 3 years, but often develop in the second or third decade of life, typically in sun-exposed areas, such as the head and neck (9, 12) (Fig. 1 D).
Management. Treatment of patients with multiple BCCs should be performed in a multidisciplinary approach, including dermatology, plastic surgery, ophthalmology and, in some cases, dentistry and otolaryngology (13). Since patients may develop multiple BCCs, the treatment should primarily include non-surgical methods, and, for patients with multiple or aggressive BCCs, treatment with a hedgehog inhibitor might be indicated (6). When the suspicion of a genodermatosis arises, clinical identification and genetic investigation is crucial in dermatological treatment and regulation. Annual check-ups should be offered and tailored to all predisposed patients. There are no perfect solutions, but prompt management of smaller BCCs can improve the cosmetic outcome.
Non-surgical methods, such as imiquimod, 5-fluorouracil (5-FU), photo-dynamic therapy (PDT) and cryotherapy, should always be considered as first-line treatments. Some patients develop hundreds of BCCs in a lifetime and every treatment should therefore be as discreet as possible.
Imiquimod is produced as a patient-applied cream and works by enhancing the innate arm of the immune system through toll-like receptor 7 (TLR7) commonly involved in pathogen recognition. Cells activated by imiquimod via TLR-7 secrete cytokines (primarily interferon-α, interleukin-6, and tumour necrosis factor-α (TNF-α)). Other cell types activated by imiquimod include natural killer cells, macrophages and B-lymphocytes. This treatment is used for actinic keratosis (AK), morbus Bowen (MB) and BCC.
5-FU cream is used for AK and BCC. It acts primarily as a thymidylate synthase inhibitor. Interrupting the action of this enzyme blocks the synthesis of thymidine, which is a nucleoside required for DNA replication. 5-FU there-fore causes rapidly dividing cancerous cells to undergo cell apoptosis as a result of thymidine depletion.
PDT is used in a variety of medical conditions, mostly localized BCC, AK and MB. A photosensitizing agent is applied on the affected skin and will be absorbed by the impaired cells. When exposed to light, the cells produce radicals and reactive oxygen species, including singlet oxygen (O2), hydroxyl radicals (•OH) and superoxide (O2−) ions. With sufficient oxidative damage, the result is targeted cell death.
For more delimited or larger tumours, curettage or surgical excision is necessary. The goal should be to maintain a cosmetically acceptable outcome while treating the underlying malignancy.
There are other rare syndromes linked to an increased risk of development of BCC. Their symptoms somewhat overlap the previously mentioned syndromes, and genetic investigation often leads to the correct diagnosis. These are listed in Table II.
Table II. List of other genodermatoses associated with basal cell carcinoma
Xeroderma pigmentosum (XP) was first described in 1874 by Hebra & Kaposi (14). XP, which means “dry pigmented skin”, is an autosomal recessive disorder with a high tumour burden. In 1968 James Cleaver described the genetic cause as a defect in DNA repair. The incidence is approximately 1 in 250,000 newborns, and it is an ultra-rare condition with a very few cases in smaller countries (15).
Molecular genetics and pathophysiology. Multiple genes have been identified as the cause of XP, all of them associated with nucleotide excision repair (NER). NER plays an essential role in the correction of UV-induced DNA damage, thereby preventing skin cancer. The diagnosis is based on family history, clinical findings and biallelic genetic mutations in the following genes: XPA, XPB, XPC, XPD, XPF, XPG or POLH. The median age at onset of BCC and SCC is approximately 8 years, more than 50 years earlier than in the general population (14). XP patients have an estimated 10,000-fold increased risk of non-melanoma skin cancer (NMSC) and a 2,000-fold increased risk of melanoma below the age of 20 years (16).
A clinically similar variant XP-V is primarily found in Europe and the USA, but unlike the other XP mutations, this variant is characterized by a failure in error-free translational synthesis past DNA photoproducts (16).
Characteristics. XP is characterized by severe photosensitivity and premature skin ageing, along with pigmentary changes and a high risk of skin malignancies, such as BCC, SCC and MM. The first indication of XP is often seen after sun-exposure, causing severe sunburn, which takes days or weeks to heal. The reaction can occur even in the first weeks of life, and is often misinterpreted as neglect or labelled wrongly as cellulitis or impetigo (17). Without sun protection, the skin ages and becomes dry, rough and atrophic (18). Small hypopigmented spots and telangiectasia can present later on. Optical photophobia is often present (Fig. 2 A, B).
Fig. 2. (A, B) Patient with xeroderma pigmentosum with hyperpigmentation of sun-exposed areas, impaired hearing and dry, atrophic skin. Rare diseases, Taylor & Francis. (C, D) Skin tumours in patients with Ferguson-Smith. (C) Upper lip. (D) A finger post self-healing. (E, F) Poikiloderma in a young patient with Rothmund-Thomson syndrome. Written permission from the patient is obtained to publish these photos. Figures A and B are published after permission from Taylor & Francis (50).
Management. Although there are individual variations, the prognosis of XP depends on minimizing sun-exposure and detecting skin changes in their earliest stages. Smaller pre-neoplasms and localized BCCs can be treated with cryotherapy or topically with 5-FU or imiquimod (16). Larger skin-lesions and SCCs should always be treated surgically, preferably with Moh’s surgery or with irrefut-able free margins due to the high risk of recurrence. XP patients also have increased risk of ocular abnormalities, along with neurological defects, and should be referred to specialized investigation at the time of diagnosis. Regular complete skin examinations are essential in early detection of precancerous skin changes and malignancies. This can improve the quality of life and, in the best case, also increase life expectancy (16).
In 1934 the Scottish dermatologist J. Ferguson-Smith described a correlation between certain symptoms in a family of Scottish miners. He depicted a condition with early onset of multiple self-healing squamous epitheliomas (MSSE) (17). In 1971 his son, geneticist M. Ferguson-Smith, ascertained the genetic inheritance as an autosomal dominant genodermatosis (19).
Molecular genetics and pathophysiology. The genetic mutation was shown, in 2005, to reside in the TGFBR1 and TGFBR2 genes, which are both tumour suppressor genes. The mutations cause an inactivation of the TGFβ-pathway, triggering uncontrolled cell growth (20). The mechanism of spontaneous healing has yet to be clarified.
Characteristics. Patients with MSSE often show symptoms early in life, presenting skin tumours highly suspicious of malignancy. The tumours usually present within 3–4 weeks, growing from a 1–2 mm red papule to a pearly nodule with central keratosis, visually imitating a keratoacanthoma. Biopsies cannot differentiate between SCC and MSSE tumours, thus a thorough history and examination of the skin is important. The tumours spontaneously self-heal after 2–3 months leaving a small recessed scar. The tumours have a tendency to local tissue invasion, but will never metastasize (20) (Fig. 2 C, D).
Management. Surgical intervention is often performed due to the histological suspicion of SCC, and if treated early on, is a good solution. However, mutilating surgery should be avoided. PDT can be used as an alternative to excision, whereas some studies have proposed that radiotherapy might worsen the condition. This statement, however, has not been substantiated by original references and does not concur with our personal experience. Tumours, situated outside the risk areas (eyes, nose, ears and mouth) can be left to self-heal, although this should be done under regular clinical supervision. Again, the goal is to keep the patient tumour-free with the best cosmetic result.
In 1967 the British physician Edgar G. Muir noted the correlation between several keratoacanthomas and the development of multiple internal malignancies at a young age (21). One year later the American dermatologist Douglas P. Torre described the same findings at the New York Dermatologic Society (22).
Muir-Torre syndrome (MTS) is a rare autosomal dominant condition with high penetrance and variable expression, thought to be a subtype of hereditary non-polyposis colorectal cancer (HNPCC). HNPCC occurs in approximately 1:350 live births, and MTS is seen in approximately 9.2% of these (22). MTS has a male to female ratio of 3:2.
Molecular genetics and pathophysiology. MTS is caused by mutations in the MLH1, MSH2, and MSH6 genes, involved in DNA mismatch repair (23). As for other genodermatoses, MTS is caused by a defects in the DNA mismatch repair, affecting rapidly dividing cells, in particular viscera and skin. Subsequently resulting in an increased risk of NMSC and visceral malignancies.
Characteristics. Patients with MTS present with a high amount of sebaceous neoplasms: adenomas, epitheliomas, and carcinomas. Skin lesions initially present as painless, slow-growing, pink or yellow nodules, often with central umbilication or ulceration. Most of the tumours are benign, but the sebaceous carcinoma can be aggressive, resulting in local invasion and metastases. Keratoacanthomas in MTS usually display sebaceous traits and multiple keratoacanthomas should give rise to further investigation.
Patients with MTS are also prone to develop malignancies of the colon and genitourinary tract (Fig. 3 D, E). The diagnosis is based on at least one sebaceous gland tumour (adenoma, epithelioma, or carcinoma) and one internal malignancy. The mean age of onset of sebaceous neoplasms is 53 years, but it can present as early as the age of 21 years (23).
Management. Patients should be offered the same strict screening for colorectal carcinoma and other malignancies as patients with HNPCC. This includes frequent and early colonoscopies, mammograms, dermatological evaluation, and imaging of the abdomen and pelvis (24). Treatment of skin malignancies consists of oral isotretinoin, which has been found to prevent tumour development (24). Tumour excision or curettage is often needed for curative tumour treatment
Fig. 3. (A, B) A 31-year-old man with keratitis-ichthyosis-deafness (KID) syndrome, alopecia and keratosis. (C) Mibelli-type hereditary porokeratosis of the abdomen. (D, E) Pale, keratotic nodule on the columella of a patient with Muir-Torre syndrome. Written permission from the patient is obtained to publish these photos.
In 1893 the Italian dermatologist Vittorio Mibelli published a case report of a young man, “age 21 [years], unmarried, of a well-to-do family of Parma” with what he termed “porokeratosis” (25). Six clinical variants of porokeratosis are now recognized: porokeratosis of Mibelli, disseminated superficial porokeratosis, disseminated superficial actinic porokeratosis, porokeratosis plantaris et palmaris disseminata, punctate porokeratosis and linear porokeratosis (25).
Molecular genetics and pathophysiology. Porokeratosis is considered a premalignant condition. All types of porokeratosis can undergo malignant transformation, most commonly, into SCC and less commonly into BCC. All forms share common features of cornoid lamella on histological examination. The lesions are thought to be due to cellular clones exhibiting varying degrees of dysplasia. Immunosuppressive diseases and drugs, burn wounds and exposure to UVR are all known contributing factors (26) (Fig. 3 C).
Characteristics. The skin lesions present as keratotic papules or annular plaques that expand centrifugally with an elevated keratotic border. When circumferentially involving the digits, it can induce pseudoainhum. Most lesions are asymptomatic, but ulcerative lesions have been described (26).
Management. Patients should be offered regular check-ups and be advised to use sun protection and avoid excessive sunlight. Upon suspicion, lesions should be biopsied. If the lesion has not undergone malignant transformation, excision is curative. Classical porokeratosis of Mibelli can be treated successfully with imiquimod cream (27). Surgical interventions and cryotherapy may be preferred when the use of topical agents is difficult or contraindicated.
In 1915 the Canadian dermatologist Frederick S. Burns first described the condition as a combination of congenital atypical ichthyosiform erythrokeratoderma, palmoplantar keratosis, and sensorineural hearing loss in a 16-year-old boy. In 1981 Skinner et al. reviewed 18 affected patients and proposed the name keratitis-ichthyosis-deafness (KID) syndrome to describe the 3 main symptoms (28). It is a rare congenital disorder of ectoderm with approximately 100 reported cases in the literature (29).
Molecular genetics and pathophysiology. KID syndrome belongs to the connexin disorders caused by heterozygous missense mutations in the connexin-26 gene, GJB2, or the connexin-30 gene, GJB6 (29). GJB2 and BJB6 are gap junction proteins expressed in ectoderm-derived epithelia of the inner ear, cornea and epidermis, which explain the constellation of pathological findings.
Characteristics. KID syndrome is characterized by deafness, erythroderma, hyperkeratotic plaques and often keratitis. Alopecia and an increased susceptibility to infections, viral, bacterial and fungal are also common. Patients are sometimes seen with a follicular occlusion triad (dissecting cellulitis of the scalp, cystic acne, and hidradenitis suppurativa) along with follicular tumours and SCC (Fig. 3 A, B).
Management. The essential issue is early diagnosis of infections and NMSC. Lifelong follow-up is recommended to assure early diagnosis of malignant tumours, especially SCC of the hyperkeratotic skin and mucosa. Systemic retinoids can reduce the hyperkeratosis and may reduce the incidence of skin cancer (30).
Rothmund-Thomson syndrome (RTS) was first described in 1868 by the German ophthalmologist Rothmund as a combination of poikiloderma, growth retardation and juvenile cataract (31). In 1936 the British dermatologist Thomson added 3 similar cases. There is an increased risk of osteosarcoma and this diagnosis should always be considered when encountering a patient with osteosarcoma, poikiloderma and NMSC.
Molecular genetics and pathophysiology. Two types of RTS have been proposed based on the clinical presentation and possible genetic mutation in the RECQL4 gene (31). It encodes for the RECQ helicase, which is responsible for correcting double-stranded DNA breaks. The loss of this gene leads to an accumulation of unrepaired DNA damage. RTS-I is an autosomal recessive heterogeneous disorder with unknown aetiology. In RTS-II there is a homozygous or compound heterozygous defect in RECQL4. The condition is considered very rare and only approximately 300 patients have been recorded in medical literature (32).
Characteristics. The severity and amount of symptoms displayed in individual patients are highly variable, but the skin, hair, nails and teeth are first to alter. The hair is often thin, brittle and sparse, and the nails are typically dystrophic with pachyonychia as a frequent sign. Dental aberrations include microdontia, rudimentary or hypoplastic teeth and disorders of dental breakthrough. A diagnostic hallmark is the erythematous, oedematous, blistering facial rash often acquired between the age of 3 and 6 months, later involving the extremities and buttocks. The rash eventually reaches a chronic phase of poikiloderma and lesions of hypo- and hyperpigmentation, telangiectatic vessels and punctate atrophy persisting throughout life (32).
The mildest variant is RTS-I, characterized by poikiloderma, juvenile cataract and ectodermal dysplasia (Fig. 2 E, F). RTS-II also causes poikiloderma, congenital bone defects, increased risk of osteosarcoma in childhood and SCC in young adults (33). Growth deficiency and skeletal defects are the second major criteria, almost exclusively seen in RTS-II as a result of the RECQL4 mutation. X-rays can be helpful to visualize defects and detect more subtle anomalies, such as radial aplasia or hypoplasia, osteopaenia, patellar ossification defects and destructive bone lesions (32).
Management. All patients diagnosed with RTS should be managed by a multidisciplinary team, including a dermatologist, an oncologist, an ophthalmologist and an orthopaedic surgeon. Patients should be offered life-long follow-up, including regular skin examinations to screen for SCC/BCC, and advice with regard to sun care. With regular screening and treatment, these patients have the same lifespan expectancy as the background population (33).
Bloom syndrome (congenital telangiectatic erythema, BS) is a rare autosomal recessive disorder. David Bloom described the condition in 1954 in a series of patients with telangiectatic facial erythema and dwarfism (34). The overall prevalence is unknown, but in the Ashkenazi Jewish population it is estimated to be approximately 1 in 48,000 live births. There seems to be a slight majority of males (35).
Molecular genetics and pathophysiology. The cause is believed to be a mutation in the BML gene, controlling the enzyme RecQL3 responsible for restoring malfunctioning replication forks during DNA replication. This defect consequently causes genetic instability due to patho-logical DNA exchanges between parallel chromatids, which may lead to malignancy (36). This may cause skin cancer, most often SCC, but also other malignancies of the upper and lower gastrointestinal and urinary tract.
Characteristics. Patients with BS are often characterized by severe growth retardation, high-pitched voices and reduced subcutaneous layer of fat, causing their muscles to appear more prominent. The patients often seek medical assistance due to recurrent infections, diabetes, chronic pulmonary disease and a predisposition to cancer development. Dermatological features include severe photosensitivity, poikiloderma and erythematous telangiectasia. SCC accounts for 14% of all tumours in patients with BS with a mean age at onset of 31.8 years (35) (Fig. 4 A, B).
Management. Patients are advised to avoid direct sun exposure and they should also be regularly screened for skin tumours. As these patients are highly susceptible to DNA damaging therapies, the use of radiation and alkylating drugs should be kept to a minimum. Dermatologists have an important role in finding and diagnosing these patients at an early stage. Although the condition is rare, it should always be considered when encountering a patient with poikiloderma, photosensitivity and multiple skin carcinomas (36).
Fig. 4. (A, B) A 3-year-old girl with Bloom syndrome. Sparse brittle hair and café-au-lait spots. (C) Male patient with epidermodysplasia verruciformis and squamous cell carcinoma of the scalp. (D) A 2-year-old child with epidermodysplasia verruciformis and plane wart-like or pityriasis versicolor-like skin lesions. Written permission from the patient is obtained to publish these photos.
This condition was first described by Lewandowsky & Lutz in 1922, who observed a correlation between infection and the development of skin malignancies. Patients present with plane wart-like or pityriasis versicolor-like skin lesions, sometimes already in childhood (37). Epidermodysplasia verruciformis (EV) is considered a very rare genodermatosis and has an estimated incidence of 1 in 1,000,000 (38).
Molecular genetics and pathophysiology. EV is an autosomal recessive skin disorder caused by mutations in the transmembrane channel TMC6/EVER1 or TMC8/EVER2 gene, making the patient extraordinarily susceptible to human papilloma virus (HPV) infections. The precise mechanism of pathology has yet to be discovered, but ultimately the patients develop HPV-induced precancerous lesions and SCCs, especially in sun-exposed areas. Approximately 90% of all cutaneous SCCs test positive for HPV 5 or 8, not included in the general HPV-vaccine (Gardasil 9 or Cervarix) (39).
An acquired form of EV has been described both in children and adults, and is primarily associated with HIV infection, but patients in immunosuppressive
therapy may also develop a clinical picture like that of EV (37). These patients also test positive for HPV 5 and 8 infections, but genetic investigations show no genetic mutations.
Characteristics. The first symptoms often develop during infancy as scaly reddish skin lesions resembling verruca plana, red-brown plaques and pityriasis versicolor-like elements on the chest and abdomen. Patients subsequently develop precancerous lesions on sun-exposed areas, which, if left untreated, often undergo malignant transformation and become invasive SCC. This will typically start in the second or third decade of life (38) (Fig. 4 C, D).
Management. As for other genodermatoses, there are currently no curative treatment options. Early detection of precancerous elements can be treated with cryotherapy. Regular skin check-ups could help prevent malignant development or at least minimize the amount of malignancies. Imiquimod, 5-FU and PDT have been used in patients with epidermodysplasia verruciformis, especially in relation to verruca plana, and retinoids have also shown promising results (38). HPV vaccination should be considered, especially in younger patients, although the currently available vaccines are too narrow in their spectrum and do not target HPV 5 or 8 (39).
Familial atypical multiple mole melanoma (FAMMM) syndrome is an autosomal dominant genodermatosis characterized by multiple melanocytic naevi, usually more than 50, and a family history of cutaneous malignant melanoma (CMM). The first case of FAMMM was reported in 1817 by William Norris (40). He described a 59-year-old man with melanoma, multiple moles and family history of this constellation. In 1968 Lynch & Krush (41) observed the same features in a family, but in this case along with an increase in pancreatic cancer. CMM is considered the most dangerous skin cancer if not detected and treated in its earliest stages. In developed countries, CMM is the sixth most common cancer, accounting for 47,000 deaths worldwide annually (42).
Molecular genetics and pathophysiology. In the 1990s the first gene mutation of FAMMM was discovered on the P16/P16INK4A gene, now known as CDKN2A (42). It is located on chromosome 9p21.3 and encodes for 2 proteins, P16 and P14ARF. In short, inactivation of the CDKN2A gene disturbs the TP53 tumour-suppressor pathways and thereby enhances proliferation and reduces apoptosis, increasing the risk of malignant transformation (43). The mutation has a reduced penetrance and a geographically variable expressivity. In 2003 Hayward described the correlation between mutations in the gene coding for cyclin-dependent kinase 4 (CDK4) and hereditary MM (44). This mutation makes CDK4 insensitive to inhibition by the protein P16, and the genetic outcome is similar to that of CDKN2A-mutations. Germline mutations of CDK4 are, however, considered very rare in FAMMM (44).
It is estimated that approximately 5–12% of all malignant melanomas are hereditary, and approximately 40% of these are caused by CDKN2A mutations. The majority of familial MM is therefore either caused by an unknown mutation or has no genetic mutation and is probably a result of a shared sun exposure and susceptible skin types (42).
Characteristics. FAMMM is a clinical diagnosis based on objective findings and only supported by genetic testing. The diagnostic criteria are shown in Table III. Most FAMMM patients have a familial history of MM, but are often not aware of their own increased risk. CMM is commonly detected between the second and third decade of life and patients frequently experience more than 1 melanoma in their lifetime (41) (Fig. 5).
Management. Dermatological screening for CMM in FAMMM patients should be offered at an early age, with a baseline complete skin examination including scalp, oral mucosa, genital area and nails, given that CMM can progress even in the early teens. Exact evaluation of naevi can be difficult even with regular dermoscopy; consequently other objective measurements, such as digital dermoscopy and computer image analysis, have been introduced to reduce diagnostic errors. Thorough counselling on sun-exposure and protection is essential in the prophylactic treatment of this patient group. Treatment of verified MM should follow national guidelines with regards to excision margins, sentinel node diagnostics, PET/CT screenings, etc. (45).
Table III. Diagnostic criteria of familial atypical multiple mole melanoma
Fig. 5. A 36-year-old man presenting with multiple naevi due to familial atypical multiple mole melanoma syndrome.
Other melanoma-predisposing gene mutations have been revealed, the most prominent of them being the BRCA1-associated protein-1 (BAP-1). Melanocytic BAP-1 mutated atypical intradermal tumours (MBAITs) were first described as a distinct entity in 2011 by Wiesner et al. (46) . It is considered an autosomal dominant syndrome caused by germline mutations of BAP-1, characterized by a high penetrance of melanocytic neoplasms. So far 4 distinct cancers have been linked to BAP-1 mutations; CMM, uveal melanoma, malignant mesothelioma and renal cell carcinoma (47).
Molecular genetics and pathophysiology. BAP-1 was first described as a binding partner of BRCA1. Its cellular role is not fully explained, but has been proposed in the DNA damage response, as well as in regulation of apoptosis, senescence, and the cell cycle (47).
Characteristics. Patients with this syndrome typically develop skin-coloured or tanned, elevated tumours, measuring approximately 5 mm, in the second decade of life (48). More tumours often develop over time, but the number of tumours can vary greatly from patient to patient. Uveal melanoma is the most common malignancy in families with BAP-1 mutations with a mean age of onset of 50 years, but patients as young as 16 years have been reported (47). These patients tend to have more aggressive cancers, with higher tumour staging and a higher risk of metastasis.
Management. Annual dermatological and ophthalmological follow up is recommended, in combination with annual abdominal ultrasounds. Magnetic resonance imaging (MRI) every 2 years could be considered, as well as annual physical examinations focusing on the risk of mesothelioma.
Patients should be given the same sun-protection guidelines as patients with CDKN2A/CDK4 mutations, as previously mentioned.
Several other mutations disposing to both cancer and melanoma have been discovered recently. In particular, 3 mutations have been associated with MM; micro-phthalmia-associated transcription factor (MITF), protection of telomeres 1 (POT-1) and the telomerase reverse transcriptase (TERT) mutations (48, 49). Germline mutations in the MITF gene are thought to increase the risk of MM, as it represents a melanocytic lineage-specific transcription factor.
Both POT-1 and TERT are telomerase-controlling genes, and mutations in these can cause uncontrollable cell cycles and malignant transformations.
Other syndromes, where the association with melanoma plays a less dominant role, exist. These include xeroderma pigmentosum (described above), Cowden syndrome (PTEN mutations), Li-Fraumeni syndrome (TP53 mutations) and hereditary breast cancer (BRCA 1/2 mutations) (49).
Genodermatoses with skin cancer predispositions are, in general, very rare, and prompt treatment and referral to genetic investigation is of utmost importance. The skin is often the first organ to display symptoms, and dermatologists therefore have a greater responsibility in early examination and executing appropriate treatment plans. Extracutaneous symptoms include pancreatic and gastrointestinal cancer, bone abnormalities, teeth malformations, neurological deficits and cognitive impairment. The above-mentioned syndromes have many similarities, and are often thought of in the same context. Exact genetic investigation is therefore crucial in establishing the correct diagnosis and treatment, especially in relation to extracutaneous symptoms.


 Click to show fullsize
Click to show fullsize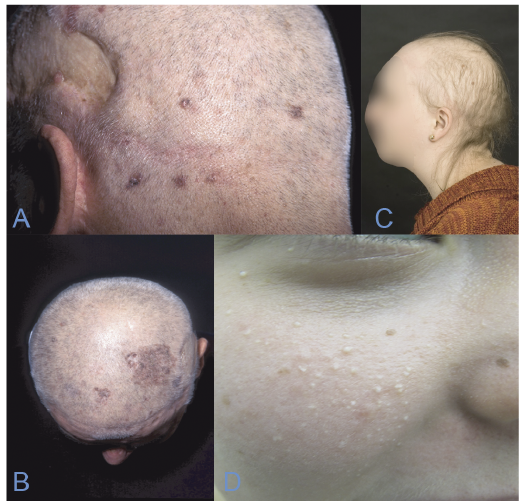 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize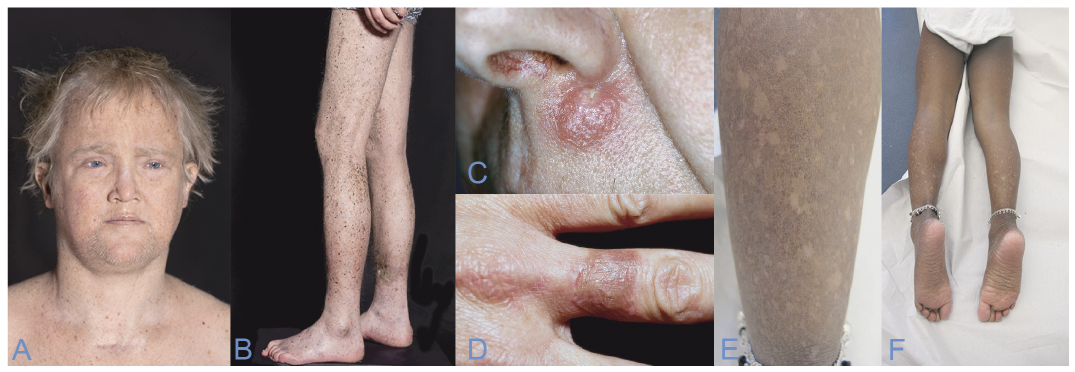 Click to show fullsize
Click to show fullsize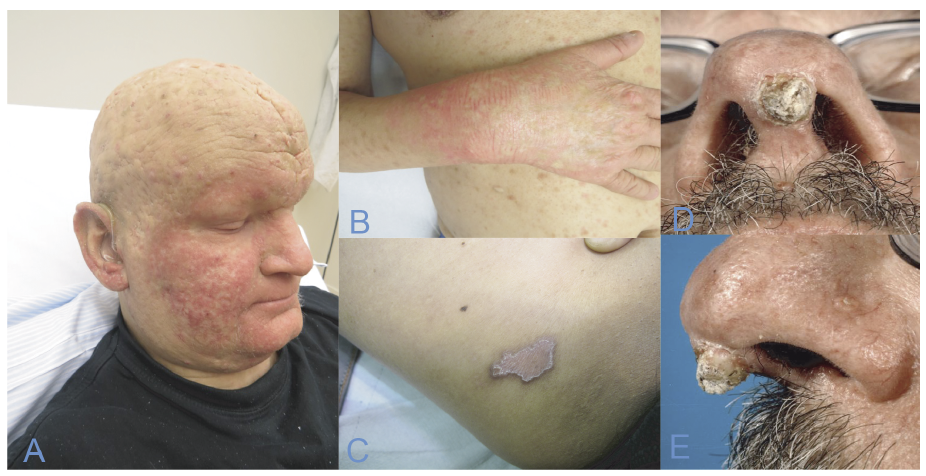 Click to show fullsize
Click to show fullsize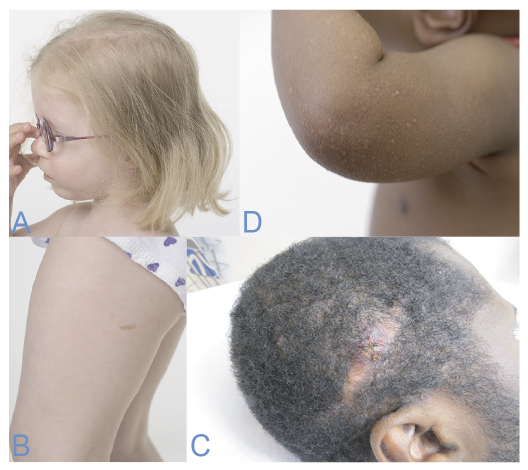 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize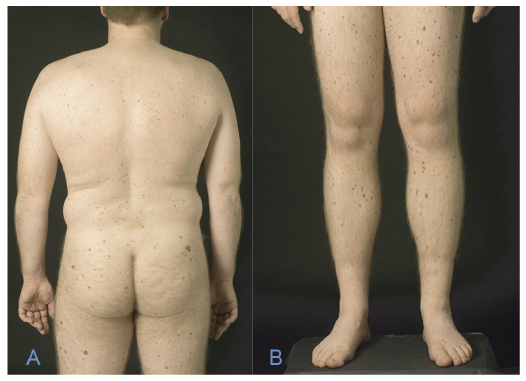 Click to show fullsize
Click to show fullsize