


1Department of Dermatology, Geisel School of Medicine, Dartmouth, 03784 West Lebanon, 2Section of Dermatology, Department of Surgery, and 3Department of Pathology and Laboratory Medicine, Dartmouth Hitchcock Medical Center, Dartmouth, USA. E-mail: catherine.m.baker.med@dartmouth.edu
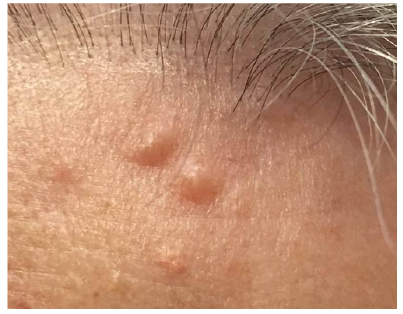
A 66-year-old woman presented to dermatology with longstanding asymptomatic firm to rubbery smooth pink papules on her forehead, posterior neck, and upper back (Fig. 1). Biopsy of one of these lesions showed a circumscribed dermal nodule comprised of bland, intersecting smooth muscle fascicles (Fig. 2). Further history revealed that the patient had undergone hysterectomy at age 34 for uterine fibroids. The patient’s mother, two daughters, and maternal aunt had also undergone hysterectomy for fibroids. Her mother, daughter, and maternal cousin had similar skin lesions.
What is your diagnosis? See next page for answer.
Fig. 1. Pair of 4-mm pink papules on forehead.
Fig. 2. Punch biopsy of a trunk papule revealed a circumscribed superficial dermal nodule comprised of intersecting fascicles of smooth muscle (H&E, original magnification 20x).
Acta Derm Venereol
Diagnosis: Pilar leiomyoma in a patient with hereditary leiomyomatosis and renal cell carcinoma
The patient was referred for genetic testing which revealed a heterozygous mutation in the fumarate hydratase (FH) gene (variant c.1293 delA). This result confirmed the diagnosis of hereditary leiomyomatosis and renal cell carcinoma (HLRCC), also known as Reed Syndrome. Further imaging with abdominal MRI revealed benign bilateral simple renal cysts – an entity that is also more prevalent in individuals with HLRCC – but no findings worrisome for renal cell carcinoma (RCC) were identified (1). Both of the patient’s daughters and one of her maternal cousins also underwent genetic testing and were found to have heterozygous mutations in the FH gene (Fig. 3). She was advised to contact her brother and remaining maternal cousins to suggest that they also be screened. No one in the family has been diagnosed with RCC.
Fig. 3. Pedigree of family of proband. Red square indicates + Fumarate Hydratase (FH) mutation, blue square indicates cutaneous leiomyomata, green square indicates uterine leiomyomata, and yellow square indicates that the patient was not tested for a FH mutation. Deceased patients are crossed.
HLRCC is a rare autosomal dominant disorder characterized by highly penetrant mutations in the FH gene on chromosome 1q42.3–43 (2). The product of the FH gene codes for the fumarase enzyme, which converts fumarate to malate in the citric acid cycle. For reasons that are poorly understood, cells that undergo loss of heterozygosity for the FH gene are at risk for undergoing tumorigenesis. It is hypothesized that cells with minimal fumarase activity accumulate excess fumarate, which stabilizes hypoxia-inducible factor (HIF) and causes cells to favour anaerobic metabolic pathways that are suitable for tumour growth (1, 3).
The exact incidence of HLRCC is unknown, but approximately 300 families with the disease have been identified (4). Individuals with HLRCC present with cutaneous leiomyomata, uterine leiomyomata, and less commonly renal tumors. Cutaneous leiomyomata generally develop in the second to third decades as multiple smooth pink, slightly firm, and sometimes painful papules arranged either singly, in groups or clusters, or in a segmental or generalized pattern. Cutaneous leiomyomata usually arise from the arrector pili muscle of the pilosebaceous unit and are therefore classified as pilar leiomyomata. They also rarely arise as genital leiomyomata from the tunica dartos in the genitals or nipple, or as angioleiomyomata from vascular smooth muscle (1). Severely symptomatic uterine leiomyomata are found in nearly all women with HLRCC and are characterized by their number, size, and early onset, usually in the third and fourth decades (1, 4). Importantly, both cutaneous and uterine leiomyomata have been reported to transform into leiomyosarcoma and therefore warrant close microscopic examination and follow–up. Lastly, the incidence of RCC in HLRCC is significantly increased compared to the general population, affecting 10–16% of individuals with the disease (5). Unfortunately, RCCs occurring in HLRCC are usually of the aggressive type II papillary type, but other tubulo-papillary and collecting-duct RCCs have also been reported in this syndrome (4).
Though diagnosis of HLRCC is strongly suggested by the presence of multiple biopsy-confirmed cutaneous leiomyomata, official diagnosis of HLRCC requires confirmation of a mutation in the FH gene. Therefore, individuals who present with either multiple cutaneous leiomyomata, a single leiomyoma and family history of HLRCC, or severe fibroids before age 40 should be sent for genetic testing so that appropriate screening regimens can be initiated (1). Current recommendations suggest that individuals with confirmed HLRCC undergo i) annual full skin exam to evaluate for the development of leiomyosarcoma, ii) annual abdominal MRI to evaluate for RCC, and iii) annual gynecologic exam with or without imaging to evaluate for fibroids and possible leiomyosarcoma (1, 6). Family members of an individual found to have a germ line FH mutation should undergo genetic testing in childhood so that annual screening for renal cancer can be started at approximately 8 years of age (3).
This case highlights the importance of recognizing the characteristic cutaneous leiomyomas of HLRCC, even when subtle in presentation. Early confirmation of this condition with genetic testing enables initiation of life-saving renal and skin cancer screening for patients and their family members.


 Click to show fullsize
Click to show fullsize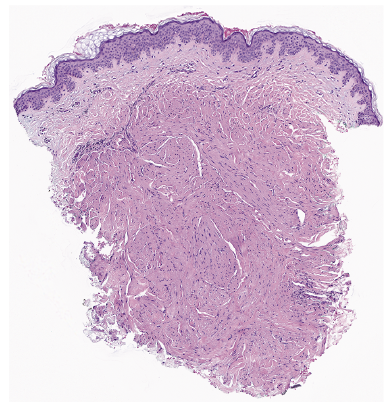 Click to show fullsize
Click to show fullsize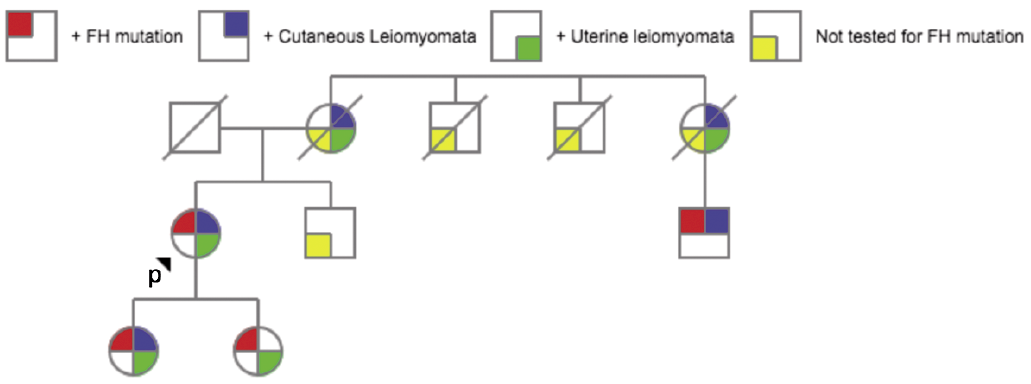 Click to show fullsize
Click to show fullsize