


Departments of 1Dermatology, 2Human Genetics, 3Pathology, and 4Pediatric Neurology, Amalia Children’s Hospital, Donders Institute for Brain, Cognition and Behavior, Radboud University Medical Center, PO Box 9101, NL-6500 HB, Nijmegen, The Netherlands. E-mail: renee.richters@radboudumc.nl
Accepted Oct 18, 2019; Epub ahead of print Oct 21, 2019
Acta Derm Venereol 2020; 100: adv00103
Oculoectodermal syndrome (OES) and encephalocraniocutaneous lipomatosis (ECL) are rare overlapping syndromes, caused by mutations of different genes in the RAS-MAPK pathway. Symptoms defining these syndromes are ocular abnormalities, such as dermoid cysts, and ipsilateral malformations of the central nervous system. We report here an exceptional case of a 3-day-old boy, who presented with a congenital hairless area covered with blister-like cutaneous lesions. Instead of known variations of RAS-MAPK genes, HRAS, KRAS and FGFR1, in the current case an NRAS variant was found. This case is the first to describe an NRAS mutation as the cause of these syndromes, and illustrates that genotypes and phenotypes in the RAS-MAPK pathway strongly overlap. The prominent bullae in this case have not been described previously in oculoectodermal syndrome and encephalocraniocutaneous lipomatosis.
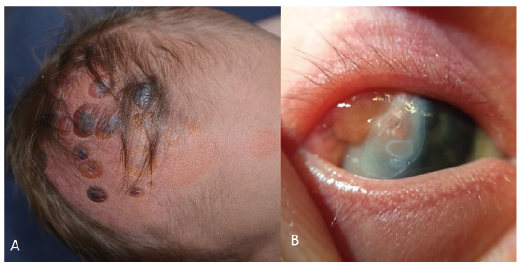
A 3-day-old boy was referred to our outpatient clinic with a congenital hairless area on the scalp covered with bullae. In addition to these cutaneous lesions, he had had nodular lesions on the sclera of the right eye since birth and hypospadias. He was born to non-consanguineous healthy parents, after an uncomplicated pregnancy of 36 weeks and 6 days due to premature rupture of membranes, and in good health. There was no family history of similar skin lesions or autoimmune conditions. Physical examination revealed a parieto-frontal yellow to erythematous atrophic hairless macule on the right side of the scalp, with multiple nummular sharply defined brown-to-violet firm bullae (Fig. 1). Overall, examination did not reveal any other cutaneous or mucosal abnormalities. Microbial culture and PCR for herpes in the fluid of one of the bullae, were negative.
Since there was no progression of the bullae and the child did not seem to suffer from the cutaneous lesions, it was decided, in agreement with the parents, to monitor the course of his condition under a diagnosis of congenital focal skin aplasia. Referral to a paediatrician did not reveal any additional abnormalities. The lesion on the right eye was diagnosed by an ophthalmologist as a lipodermoid cyst. Re-examination after one month found that a subcutaneous swelling was visible in the area of the bullae, whereas the position and aspect of the bullae had not changed. Ultrasonography of the lesion and the underlying scalp revealed focal aplasia of bone in the area of the increased subcutaneous swelling.
Fig. 1. Photographs of the patient at the age of 3 days, showing: (a) erythematous atrophic hairless macules with multiple nummular sharply defined yellow-to-violet firm bullae; and (b) lipodermoid cyst of the right eye.
The patient was referred to the paediatric neurologist, who found no clinical neurological abnormalities at that time, but requested cerebral magnetic resonance imaging (MRI) due to suspicion of a neurocutaneous syndrome. MRI revealed asymmetrical hemispheres with a smaller right hemisphere volume, polymicrogyri of the right parietal cortex, increased subarachnoidal space, arachnoidal cystic configuration at the right temporal fossa, a choroid plexus cyst in the right ventricle and small lacunar changes (Fig. 2). No intracranial lipomatous abnormalities were found.
Genomic DNA was isolated from affected skin tissue, obtained via a 4-mm skin punch biopsy, and from blood lymphocytes, in order to identify gene variations causing ECL and OES. The coding exons and intron-exon boundaries of the FGFR1 gene (NM_023110) were sequenced either by Sanger or ion semiconductor sequencing. Copy number variation in FGFR1 gene was investigated by MLPA analysis (P133-C1 kit, MCR-Holland, The Netherlands). In addition, single molecular Molecular Inversion Probe (smMIP) based library preparation was performed followed by NGS analysis on a NextSeq500 instrument (Illumina, San Diego, CA, USA) including unique molecule identifiers (UMIs), with an analytical sensitivity, as described by Eijkelenboom et al. in 2016 (1). The panel included frequently mutated positions in AKT1, BRAF, CTNNB1, CXCR4, EGFR, ERBB2, EZH2, GNA11, GNAQ, GNAS, H3F3A, H3F3B, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, MYD88, NRAS, PDGFRA, PIK3CA and SF3B1. No unclassified or pathogenic variant, exon deletion or duplication was identified in the FGFR1 gene in DNA isolated from lymphocytes or affected skin tissue. However, smMIP panel analysis detected an NRAS variant c.182A>G (p.(Gln61Arg)) at 16% variant allele frequency in affected skin tissue, while no suspected variants were identified in the other investigated genes, including KRAS. The variant in the NRAS gene was not identified in DNA isolated from circulating lymphocytes. Based on the clinical phenotype and the NRAS mutation, a diagnosis of OES/ECL was made.
Fig. 2. Axial T2-weighted magnetic resonance images of the brain showing: (a) a large arachnoidal cyst at the right temporal fossa; and (b) a small volume of the right hemisphere, polymicrogyri of the right parietal cortex, and increased subarachnoidal spaces.
OES and ECL are rare syndromes; only 20 and 50 cases of OES and ECL, respectively, have been reported (2, 3). These syndromes strongly overlap with respect to phenotype and genotype. Recently, OES has been considered a subtype of ECL (2, 4, 5). Ocular abnormalities, such as dermoid cysts, and ipsilateral malformations of the central nervous system contribute to the triad defining the syndromes. The diagnostic criteria for ECL were proposed by Hunter (6), including, amongst others, the following major criteria: (i) eye: ocular choristoma; (ii) skin: naevus psiloliparus (a mesodermal naevus of the scalp characterized by absence or paucity of hair and an excessive amount of fatty tissue); congenital craniofacial lipomas; multiple small craniofacial skin tags, skin aplasia or hypoplasia, possible findings of naevus psiloliparus with lipomas; (iii) brain: intracranial lipoma, intracranial calcification and arachnoid cysts. In OES, the eye and skin symptoms are also seen frequently, including epibulbar dermoids, focal aplastic skin defects and non-scarring alopecia of the scalp. A major difference is the lack of intracranial lipomatosis (5, 6). In the current case, intracranial lipomatosis could not be confirmed, suggesting the diagnosis of OES, which can be considered a subtype of ECL, according to recent literature (2, 6).
OES and ECL were found to be caused by mutations of different genes in the RAS-MAPK pathway (HRAS, KRAS and FGFR1), resulting in these overlapping phenotypes (2, 4, 5). Local involvement of the skin implies an underlying mosaicism. Mechanisms such as ectodermal dysgenesis and mosaicism are thought to be responsible for a mutated autosomal gene leading to mesenchymal tumours and vasculogenesis (7). In 2016, Bennett et al. (8), discovered mutations in FGFR1 in ECL, and therefore, this was the first analysis that we requested in order to find a mutation in this case. Later in 2016, ECL was linked to mutations in the RAS-MAPK pathway (8, 9). In 2015, Peacock et al. (3) also discovered mutations in the KRAS gene in 2 cases of OES, affecting p.Leu19Phe and p.Gly13Asp. Recently, these mutations have been shown in 4 other cases, affecting codon 146 of the KRAS gene (p.Ala146Val and p.Ala146Thr), confirming the role of the RAS-MAPK pathway (4, 10).
An NRAS variant in a mosaic pattern was found instead of a mutation in FGFR1 or KRAS. NRAS variants are known to play a role in congenital giant melanocytic naevi, Schimmelpenning-Feuerstein-Mims syndrome and Noonan syndrome (11). These variants have not been de-scribed previously in relation to OES/ECL. NRAS is also a RAS-MAPK gene and, therefore, it may not be surprising that mutations in NRAS also can lead to this phenotype. In 2017, Altmuller et al. (11) described phenotype and genotype spectra of NRAS variants, describing a case with ectodermal dysplasia and intracerebral arachnoid cyst and subcutaneous oedema. Next to this new variant, this is the first case in which blister-like cutaneous lesions have been described in a patient with OES/ECL. However, bullae have been described frequently in aplasia cutis in the context of syndromic or non-syndromic cases in general (12). NRAS mutations have not been described in relation to bullae. Given the convergent localization, however, we hypothesize that the bullae are part of OES/ECL, and therefore present a new clinical aspect of this uncommon disease.
In conclusion, this case is the first to describe an NRAS mutation as the cause of OES/ECL. It illustrates that genotypes and phenotypes in the RAS-MAPK pathway strongly overlap, and that in addition to HRAS and KRAS, NRAS mutations can also lead to OES/ECL.


 Click to show fullsize
Click to show fullsize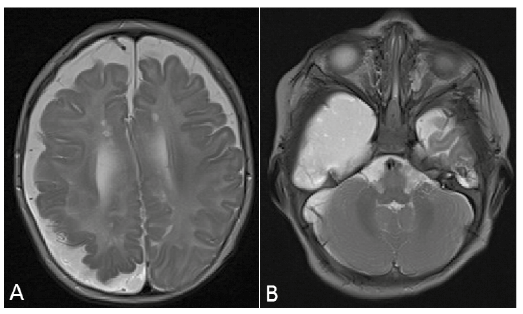 Click to show fullsize
Click to show fullsize