


1Department of Dermatology and 7Department of Pathology, Hôpital Cochin, Assistance Publique des Hôpitaux de Paris, AP-HP Centre-Université de Paris, 2Université de Paris, 3Department of Hematology and Bone Marrow Transplantation, Hôpital Saint-Louis, 4Department of Dermatology, 5Department of Pathology, Hôpital Saint Louis, Assistance Publique des Hôpitaux de Paris, AP-HP Nord-Université de Paris, and 6Department of Pediatric Hematology and Oncology, Hôpital Trousseau, Assistance Publique des Hôpitaux de Paris, AP-HP Est-Université de Paris, Paris, France. *E-mail: coralie.lheure@aphp.fr
Accepted Oct 15, 2020; Epub ahead of print Oct 19, 2020
Acta Derm Venereol 2020; 100: adv00338.
doi: 10.2340/00015555-3671
Pyoderma gangrenosum (PG) is a neutrophilic dermatosis (ND) characterized by a chronic ulcerating skin condition that appears to be immune mediated. Recent diagnostic criteria have been proposed for PG as a result of a Delphi consensus exercise using the RAND Health Care/University of California at Los Angeles Appropriateness Method (1). The cause of PG remains unknown, but up to 50% of cases occur in association with multiple systemic diseases, such as inflammatory bowel disease, rheumatological conditions, or malignant haematological disorders, in particular acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS) (2, 3). A clonal link between malignant myeloid cells and skin-infiltrating neutrophils has been described recently in ND associated with AML and MDS (4). Fanconi anaemia (FA) is a rare autosomal recessive bone marrow failure syndrome due to genetic defects of a DNA repair pathway. The major complications of FA are aplastic anaemia, AML, MDS and specific solid tumours. We describe here 2 adult patients with long-standing FA diagnoses who developed PG, revealing an acceleration of their myeloid disorder. To our knowledge only 2 similar cases have been reported previously (5, 6).
Case one. The first patient was a 35-year-old woman diagnosed with FA at the age of 5 years. She developed progressive thrombocytopaenia at the age of 12 years, and received androgens from the age of 20 years, with a long-term partial response. Treatment was stopped at the age of 27 years, while she was still responding. Her last cytological bone marrow examination, at the age of 25 years, was normal, as was her bone marrow cytogenetic analysis.
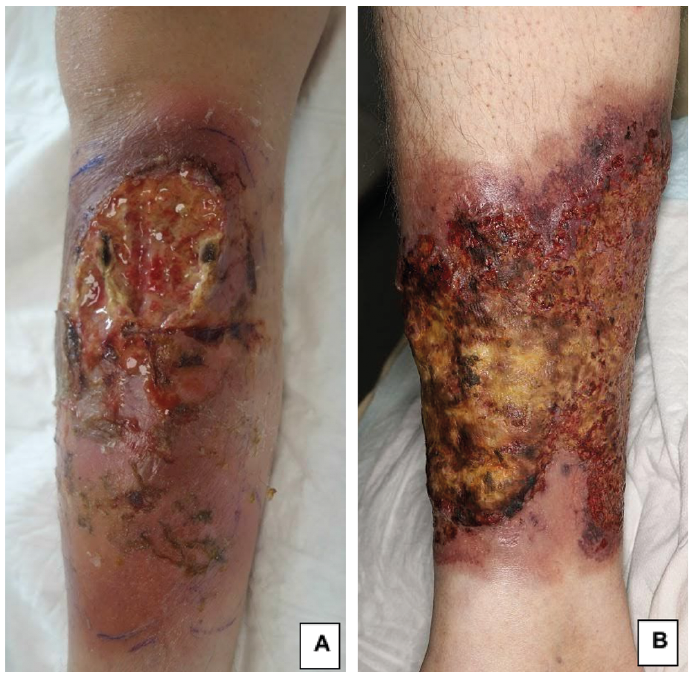
At the age of 34 years, the patient developed a rapidly growing purulent ulceration on her right leg (Fig. 1A), associated with fever. She was initially treated with broad-spectrum antibiotics and abscess drainage, without improvement. Microbiological investigations detected no bacteria, mycobacteria, fungi or parasites. Histological analysis of a skin biopsy specimen showed prominent neutrophilic dermal infiltrate and micro-abscesses. A diagnosis of PG was retained. According to the Delphi consensus, the patient fulfilled the major criteria and 5/8 minor criteria (1). Treatment with prednisone (0.75 mg/kg per day) was initiated. The fever disappeared within 48 h and the cutaneous lesions gradually improved.
Bone marrow aspiration was performed while blood cells counts were still in the normal range. Marked signs of erythroid and megakaryocytic dysplasia were found, and cytogenetic studies showed a t(6;10) translocation and a 7q addition, confirmed by fluorescence in situ hybridization (FISH) analysis. These results reflected clonal evolution towards a high-risk myelodysplastic syndrome.
The PG lesions healed completely with corticosteroid treatment, but a relapse occurred 3 months later, while the patient was waiting for a matching donor for allogeneic stem cell transplantation. The dose of steroids administered was therefore increased.
Case two. The second patient was diagnosed with FA at the age of 10 years. She had moderate anaemia and thrombocytopaenia from the age of 26 years onwards. At the age of 32 years, she developed PG on her right leg, characterized by an ulcerated and purulent lesion (Fig. 1B). A biopsy of the ulcer edge showed a neutrophilic infiltrate, fulfilling the major diagnostic criteria of an ulcerative PG, as well as 5/8 minor criteria. A combination of topical and oral corticosteroids was effective, and treatment was stopped after one year. A bone marrow smear revealed MDS without excess blasts, and cytogenetic examination revealed an addition to chromosome 2 in most mitoses. This patient had never undergone transfusion or any specific haematological treatment.
Fig. 1. Necrotic and ulcerative lesions of pyoderma gangrenosum in (A) patient 1 and (B) patient 2.
One year later, the lesion relapsed and became deep and purulent. Anaemia and the thrombocytopaenia worsened, as shown by biological parameters, and the patient required several blood transfusions. Treatment with 5 mg/kg infliximab was initiated, without improvement. Pancytopaenia progressed further, necessitating several blood transfusions. Cytological examinations of bone marrow revealed persistent features of dysplasia, without blasts. Cytogenetic studies demonstrated the presence of additional abnormalities, including partial trisomy for chromosome 8q, a feature correlated with a high risk of progression to AML. PG in this patient followed a chronic course, over a period of 5 years and still requires a high dose of oral steroids (> 30 mg/day), with adverse effects, including osteoporosis in particular. A partial improvement of PG was obtained by replacing infliximab with etanercept, as the patient developed severe infections on infliximab. She is currently on etanercept treatment and awaiting allogeneic stem cell transplantation.
The association between PG and haematological disorders is well known. The most commonly reported haematological disorder is MDS (24.4%), followed by monoclonal gammopathy of unknown significance (22.1%) and AML (11.5%). In most cases, the diagnosis of a malignant haematological disorder precedes the development of PG (7). A recent next-generation sequencing (NGS) study performed in France demonstrated a clonal origin for skin-infiltrating cells from myeloid neoplasm-associated ND. The polymorphonuclear cells infiltrating the dermis and the malignant myeloid clone were found to have a common clonal progenitor (4).
ND has rarely been described in association with FA: 11 cases of Sweet’s syndrome and 2 cases of PG (5, 6, 8). In almost all cases, the onset of Sweet’s syndrome was associated with progression to malignant haematological disease. In a series of 7 cases of FA, the development of Sweet’s syndrome was reported between the ages of 17 and 36 years (9). One case of PG occurred after the underlying malignant disease had been diagnosed (5). The other case of PG led to the discovery of the underlying FA and its progression towards MDS (6).
In both the patients described here, we consider the onset of PG to be a sign of the haematological malignancy: MDS with a high risk of progression to AML in both cases. Blood cell counts were stable at PG diagnosis, and PG revealed clonal progression in both cases. The onset of PG in these patients facilitated the early detection of additional clonal cytogenetic abnormalities associated with a high risk of progression to AML. This may facilitate the early introduction of appropriate treatment.
There are currently no guidelines for PG treatment. Corticosteroids are the most widely used treatment, in a topical, intralesional or systemic form. Other immunosuppressive or immunomodulating agents have also yielded positive results for the management of PG. In 2015, a randomized controlled trial found that infliximab was superior to placebo at 2 weeks (46% vs 6% response), with a 21% complete healing rate at 6 weeks (10). Two uncontrolled trials showed 60% and 37% healing within 4 months for canakinumab and infliximab, respectively (11, 12). However, Ben Abdallah et al. (13), in their review, did not find significant differences between response rates with infliximab, adalimumab and etanercept. Concerning calcineurin inhibitors, such as ciclosporin, one randomized controlled trial including 121 patients showed that prednisolone and ciclosporin were similar: 15–20% of patients showed complete healing at 6 weeks and 47% at 6 months (14). Other immunomodulating treatments were reported with positive effect in PG: dapsone, thalidomide, azathioprine, cyclophosphamide, methotrexate, and mycophenolate mofetil (15). Thalidomide is potentially the most interesting of these treatments, given its antiangiogenic and anti-inflammatory effects on PG lesions, and the improvement in cytopaenia reported in patients with MDS (15, 16). However, the primary curative therapy for FA-associated AML is allogeneic stem cell transplantation.
In conclusion, the onset of PG in a patient with FA should raise questions about the possibility of clonal progression of the disease and should lead to bone marrow analysis with conventional cytogenetic and FISH analysis.


 Click to show fullsize
Click to show fullsize