


1Institute of Human Genetics and 6Department of Dermatology, Medical Center – Faculty of Medicine, University of Freiburg, Freiburg, 2Department of Dermatology and Allergology, Philipp University, Marburg, Germany, 3Service de Dermatologie, Hôpital Saint-Louis, 1, avenue Claude-Vellefaux, Paris, France, 4Department of Dermatology and Venereology, Münster University Medical Center, Münster, Germany, 5Centro Integral de Dermatología, Tucumán, Argentina and 7Department of Dermatology, Venereology, and Allergology, University Hospital Würzburg, Würzburg, Germany
Porokeratoses are a heterogeneous group of keratinization disorders. For linear porokeratosis and disseminated superficial actinic porokeratosis, a heterozygous pathogenic germline variant in a mevalonate pathway gene and a postzygotic second hit mutation present in affected skin have been shown to be the pathogenetic mechanism for the development of the lesions. However, the molecular mechanism leading to development of porokeratosis plantaris, palmaris et disseminata is not known. This study analysed a cohort of 4 patients with linear porokeratosis and 3 patients with porokeratosis plantaris, palmaris et disseminata, and performed mutation analyses of DNA extracted from blood samples and skin biopsies. All of the study patients carried the heterozygous germline variant c.70+5G>A in the MVD gene. Loss of heterozygosity due to a second hit mutation was found in affected skin of 3 patients with linear porokeratosis and 2 patients with porokeratosis plantaris, palmaris et disseminata. These results suggest that porokeratosis plantaris, palmaris et disseminata shares the same pathogenetic mechanism as other porokeratosis subtypes and belongs to the phenotypic spectrum of MVD-associated porokeratosis.
Key words: mevalonate diphosphate decarboxylase gene; porokeratosis plantaris; palmaris et disseminata; linear porokeratosis; mosaicism; second hit; loss of heterozygosity.
Accpeted Jan 21, 2021; Epub ahead of print Jan 21, 2021
Acta Derm Venereol 2021; 101: adv00397.
doi: 10.2340/00015555-3753
Corr: Judith Fischer, Institute of Human Genetics, Medical Center – Faculty of Medicine, University of Freiburg, Breisacher Str. 33, DE-79106 Freiburg, Germany. E-mail: Judith.fischer@uniklinik-freiburg.de
The molecular cause of porokeratosis plantaris, palmaris et disseminata is unknown. This study found that, on a molecular level, porokeratosis plantaris, palmaris et disseminata is caused by a first mutation that is present in all cells of the body plus a second mutation found only in the skin lesions. The molecular mechanism for the development of porokeratosis plantaris, palmaris et disseminata is therefore the same as in other porokeratosis subtypes. Since the first mutation is present in all cells of the body, people with porokeratosis plantaris, palmaris et disseminata can pass on this mutation to their children, who are therefore at higher risk of also developing porokeratosis.
Porokeratoses are a heterogeneous group of keratinization disorders characterized by keratotic papules and annular plaques with raised hyperkeratotic borders (1–3). On histopathological examination, these borders are characterized by thin columns of tightly packed parakeratotic cells within the stratum corneum, the so-called cornoid lamella (1–3). Moreover, the stratum granulosum is absent beneath the parakeratotic column and there is vacuolization and, sometimes, pleomorphism and dyskeratosis of keratinocytes (4, 5). Several clinical variants, which differ in morphology and distribution of the lesions, can be distinguished, among them porokeratosis of Mibelli (PM), disseminated superficial actinic porokeratosis (DSAP), porokeratosis plantaris, palmaris et disseminata (PPPD), and all of these may show the phenomenon of linear porokeratosis (LP) (1–3).
DSAP, the most common subtype, often starts in the 3rd to 4th decade of life. Skin lesions are typically smaller than 1 cm in diameter and arise on sun-exposed areas of the extremities. The number of lesions can vary from a few to several hundred (2, 3). Classic PM usually begins in childhood as a small keratotic papule gradually enlarging to a plaque up to several centimetres in size, with a thin, elevated rim of hyperkeratosis, and central atrophy (2, 3). LP lesions follow the lines of Blaschko and develop in infancy or childhood. Depending on their size, number and distribution, the extent of LP can vary considerably, with either localized or systematized involvement (2, 3). Porokeratosis can undergo malignant transformation to squamous cell carcinoma (SCC), for which LP seems to be especially susceptible (6). PPPD is an uncommon subtype of porokeratosis, first described in 1971 by Guss et al. (7). Usually, lesions appear first on palms and soles in childhood or adolescence and may later develop on other body parts, such as the trunk and extremities (7, 8). However, cases with first lesions on the body followed by palmar and plantar lesions have also been described (9–11). Punctate porokeratosis (PP), like the PPPD subtype, presents with small papules confined to the palms or soles, resembling punctate keratoderma. Some authors assume PP to be a “forme fruste” of PPPD (12).
Pathogenic variants in genes of the mevalonate pathway (MVK (MIM *251170), PMVK (MIM *607622), MVD (MIM *603236) and FDPS (MIM *134629)) have been identified as the genetic cause of different porokeratosis subtypes, PM and DSAP, including LP (13, 14). Recently, for LP, the long-standing hypothesis of type 2 segmental mosaicism (15) was molecularly proven. Atzmony et al. (16) detected heterozygous germline mutations plus a second postzygotic mutational event leading to loss of the wild-type allele (loss of heterozygosity; LOH) or to compound heterozygosity in affected skin of patients with LP. It is notable that somatic second hit mutations in affected skin were also proven for non-segmental DSAP (17). Presumably, the second hit in the keratinocytes in LP lesions happens during embryogenesis, while, in DSAP lesions, the second hit is a postnatal event initiated, for example, by exposure to ultraviolet (UV) light (17, 18). Wei et al. (19) assigned the PPPD locus to chromosome 12q24.1–q24.2 and proposed an autosomal dominant mode of inheritance. However, the underlying gene defect and pathogenetic mechanism for this particular subtype have not yet been fully clarified.
Patients
A total of 7 patients with clinical diagnoses of different forms of porokeratosis were included in the study: 4 cases with LP from the Department of Dermatology, Venereology, and Allergology of the University Hospital Würzburg, Germany, the Department of Dermatology and Venereology of the University Medical Center in Münster, Germany, and the Centro Integral de Dermatología in Tucumán, Argentina, and 3 patients with a diagnosis of PPPD from the Department of Dermatology and Allergology of the University Hospital Marburg, Germany and the Department of Dermatology, Hôpital Saint-Louis, Paris, France. The clinical features of patient 1 have been described by Goebeler et al. (20). Informed consent was obtained from all patients.
Next generation sequencing analyses
For isolation of genomic DNA from EDTA blood or fresh skin biopsies, Maxwell 16 LEV Blood DNA Purification Kit (Promega, Fitchburg, WI, USA) was used according to the manufacturer’s instructions. For detection of pathogenic DNA variants in skin tissue next generation sequencing (NGS) was performed, either multigene panel or whole exome analyses, using an Agilent SureSelect Custom Kit or the SureSelect XT Human All Exon V7 Kit for DNA enrichment (Agilent, Santa Clara, CA, USA). Sequencing (2×150 bp, paired end) was performed using an Illumina MiSeq or NextSeq (Illumina, San Diego, CA, USA) device, respectively. Bioinformatic analyses were performed using an in-house bioinformatic pipeline (alignment: bwa version 0.7.15-r1140 against the human genome hg19; variant calling: GATK version 3.8 nightly-2017-12-06-1; annotation and variant filtering: Variant Effect Predictor (VEP) version 96, gnomAD Version 2.1 und Gemini Version 0.30.2.) and the Software SeqNext (JSI Medical Systems, Ettenheim, Germany). All exonic regions ±20 bp at exon/intron borders of the MVD gene were covered with a minimum of 10 reads.
Sanger sequencing
For analysis of DNA extracted from EDTA blood and for validation of pathogenic variants detected by NGS in DNA extracted from skin tissue, Sanger sequencing was performed. The following primers were used for PCR amplification of exon 1 + 20 bp at the exon intron boundary of the MVD gene (NM_002461.2, GRCh37.p13): forward: 5’-CGATCCCGTCCATTGGCTG-3’, reverse: 5’-TAATCGCTAATGGGCAGGCG-3’. PCR was performed using standard methods and Sanger sequencing was performed on an ABI 3500 DNA Sequencer (Applied Biosystems, Foster City, CA, USA).
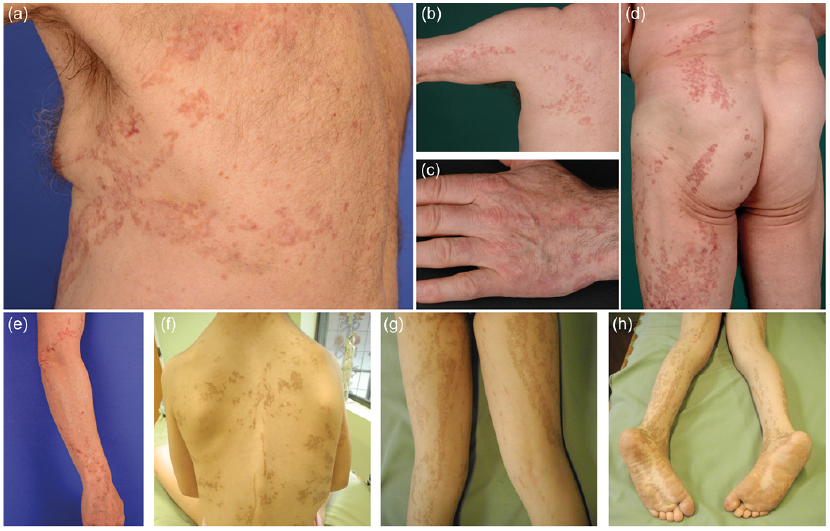
Four patients with LP and 3 with non-segmental PPPD were included in this study. Patients 1–4 (P1–P4), affected by LP, presented with either localized lesions (P1–P3) or with a systematized form of linearly arranged porokeratosis (P4). Distribution of the lesions was as follows: P1: on the left side of the thorax extending to the left arm (Fig. 1a); P2: on the left arm (Fig. 1b), hand (Fig. 1c) and left side of the trunk extending to the left leg (Fig. 1d); P3: on the right arm (Fig. 1e); and P4: on the face, trunk (Fig. 1f), legs (Fig. 1g) and feet (Fig. 1h). In patient 1, in addition to the linear lesion, disseminated porokeratotic papules were observed on the extremities. In the patients with LP the linear lesions had been noted since infancy/childhood. P2 developed SCCs in his 6th and 7th decade of life on the left thigh, the left buttock and the back of the left foot, in accordance with the observation that LP is associated with a higher risk of malignant transformation (6).
Fig. 1. Phenotype of patients with linear porokeratosis. (a) Patient 1: Porokeratotic lesions in linear arrangement on the left side of the thorax. (b, c, d) Patient 2: Linear porokeratosis on (b) the left arm, (c) the hand and (d) left side of the back and on the left thigh. (e) Patient 3: Linear porokeratosis on the right arm. (f, g, h) Patient 4: Systematized porokeratotic lesions on the (f) trunk, (g) legs, and (h) feet.
Patients with PPPD (P5–P7) had multiple small hyperkeratotic lesions on the feet (Fig. 2a, b (P5), Fig. 2c (P6), Fig. 2d (P7)) and hands (Fig. 2e (P5) and Fig. 2f (P7)), which is typical for this subtype. In addition, patients 5 and 7 had innumerable disseminated annular lesions on the trunk (Fig. 2g, h) and patient 7 also on the legs (Fig. 2i). In all patients with PPPD the lesions appeared first on the soles. The age of onset was 18 (P5), 7 (P6) and 13 years (P7), respectively. The lesions which were not on the palms and soles in patients 5 and 7 developed later in life. Patient 6 (1 of 4 daughters of patient 5) is 27 years of age and, to date, presents only plantar lesions.
Fig. 2. Phenotype of patients with porokeratosis plantaris, palmaris et disseminata. Lesions on (a) the back of the right foot of patient 5 and plantar lesions of patients (b) 5, (c) 6 and (d) 7. Lesions on the left palm of (e) patient 5 and (f) on the proximal fingers of patient 7. Disseminated porokeratotic lesions on the ventral trunk of patients (g) 5 and (h) 7 and (i) on the legs of patient 7.
In order to identify the underlying genetic causes the DNA extracted from skin biopsies and blood samples was sequenced by NGS and Sanger methods. It is notable that, in all patients irrespective of porokeratosis subtype, the heterozygous pathogenic germline variant c.70+5G>A in the MVD gene was detected in DNA isolated from blood cells (Table I). Since for LP and DSAP the pathogenetic mechanism is attributed to the combination of a heterozygous germline variant and a second hit mutation in the affected skin (16, 17), we screened for the MVD gene in both tissues of patients P1, P2 and P4, from whom also skin biopsies were available. In patient 1 no second hit mutation could be identified in the MVD gene in punch biopsies from the LP and a non-segmental DSAP lesion. However, in samples from LP lesions obtained by curettage of P1 as well as in punch biopsies from P2 and P4 the alternative A allele of the variant c.70+5G>A was detected in the MVD gene, with an allele frequency of 65%, 67% and 65% (Fig. 3a–c), respectively, suggesting a second hit mutation resulting in LOH in affected skin (Table I). Since the current study also identified the variant c.70+5G>A heterozygous in the germline in the patients with PPPD, we also checked for second hit mutations in affected skin. In 2 of the 3 patients (P5 and P7) a higher frequency of the alternative A allele of 63%, 55% (P5) and 77% (P7) was observed in plantar skin biopsies (Fig. 3d–f), indicating that loss of the wild-type allele had also occurred in PPPD lesions.
Table I. Summary of the germline (1st-hit) and postzygotic somatic (2nd-hit) pathogenic variants in the MVD gene identified in patients with linear porokeratosis and porokeratosis plantaris, palmaris et disseminata
Fig. 3. Results from the NGS and Sanger analyses of affected skin tissue. In the top part electropherograms (left side) and parts of exemplary reads (right side) of the NGS analyses of (a) patient 1, (b) patient 2, (c) patient 4, (d, e)patient 5 (d (caudal biopsy), e (cranial biopsy)) and (f) patient 7 are shown. Sanger sequencing results of the respective patients are depicted below.
In 2003, Wei et al. (19) assigned the PPPD gene locus to chromosome 12q24.1–q24.2. However, Happle (21) argued that the authors did not analyse a family affected by PPPD, but instead a family affected by PM. In line with this assumption, the PM-causing gene MVK is located on chromosome 12q24.11. The current data show that PPPD is caused by pathogenic variants in the MVD gene located on chromosome 16q24.1.
In all 7 patients in the current cohort the pathogenic heterozygous germline variant c.70+5G>A in the MVD gene was detected. This variant leads to aberrant splicing and nonsense-mediated decay (16). In the population database Genome Aggregation Database (gnomAD) the total allele frequency of this variant is estimated as 0.2%. In the European (non-Finnish) population it was detected in 401 out of 115,610 alleles (minor allele frequency of 0.003 (0.3%)), suggesting that approximately 1 per 150 individuals carries this variant. Therefore, it is assumed to be a frequent porokeratosis-associated pathogenic variant, especially in Europeans.
In one of the patients with LP no second hit mutation was detected in affected skin in material from a punch biopsy, but the pathogenic variant was detected in material obtained by curettage. It is possible that curettage is more suitable to yield specifically the affected keratinocytes with second hit mutations without a marked admixture of dermal cells, which most likely do not carry the additional mutational event (17).
The variant c.70+5G>A in the MVD gene has been detected to be heterozygous in the germline in patients affected by different porokeratosis subtypes, DSAP and PPPD (22), including patients with LP (16). Even though this variant has already been detected as heterozygous in the germline of patients with PPPD, no second hit mutation was identified in studies by Li et al. (23) and Atzmony et al. (22). Thus, the molecular explanation for the development of the lesions in this specific subtype is still lacking. The data from the current study suggest that PPPD lesions, such as DSAP and LP lesions, are also caused by a second hit mutation present in affected skin. In principle the second hit could be due to LOH (as observed in the patients with PPPD in the current study) or to compound heterozygosity (which was additionally described for non-segmental DSAP and LP) (16, 17). Kubo et al. (17) provided evidence that linear lesions are caused by one early postzygotic second hit mutation present in cells of LP lesions, whereas the disseminated lesions in DSAP are caused by different individual second hits arising in aged skin. Because PPPD, like DSAP, is characterized by disseminated lesions appearing later in life, it would be interesting to investigate whether different second hits can also be detected in PPPD lesions.
It is not known why some individuals carrying the c.70+5G>A variant develop PPPD manifesting predominantly on feet and hands, whereas others develop DSAP commonly restricted to sun-exposed areas of the extremities. Comparison of the exome data of the current patients with PPPD did not identify any further DNA variation(s) linked to the development of PPPD. Further research comparing more patients with PPPD may reveal predisposing factors for this specific and rare porokeratosis subtype.
In conclusion, PPPD belongs to the spectrum of MVD-associated porokeratoses.
ACKNOWLEDGEMENTS
The authors would like to thank all patients for taking part in this study and supporting this work. The patients in this study provided written informed consent to publication of their case details. The authors are grateful to Stefanie Kupferer, Christine Hodler, Angela Steiert, Vlada-Irina Nuhn, Hande Güler and Susanne Vidal for excellent technical support.
The study was supported by a grant from the German research foundation DFG (FI1767/3-3).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize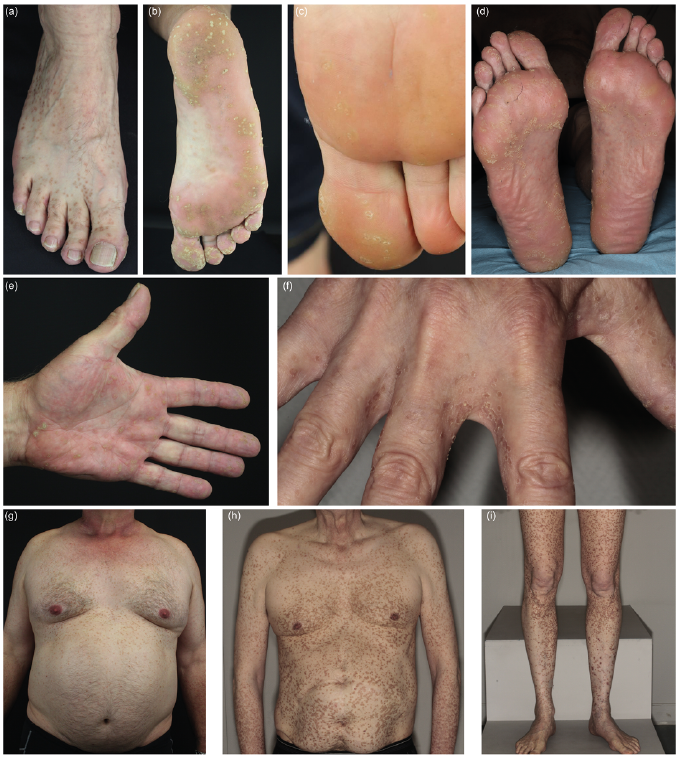 Click to show fullsize
Click to show fullsize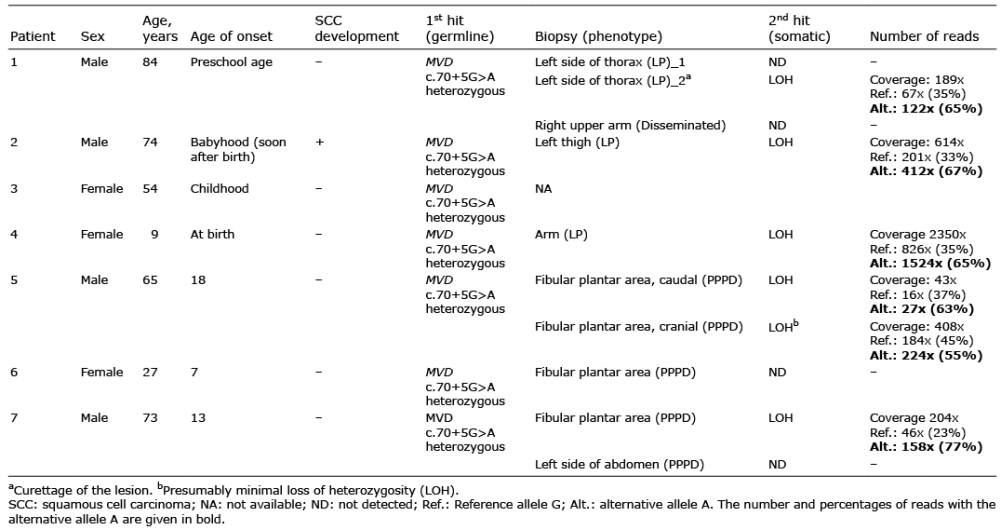 Click to show fullsize
Click to show fullsize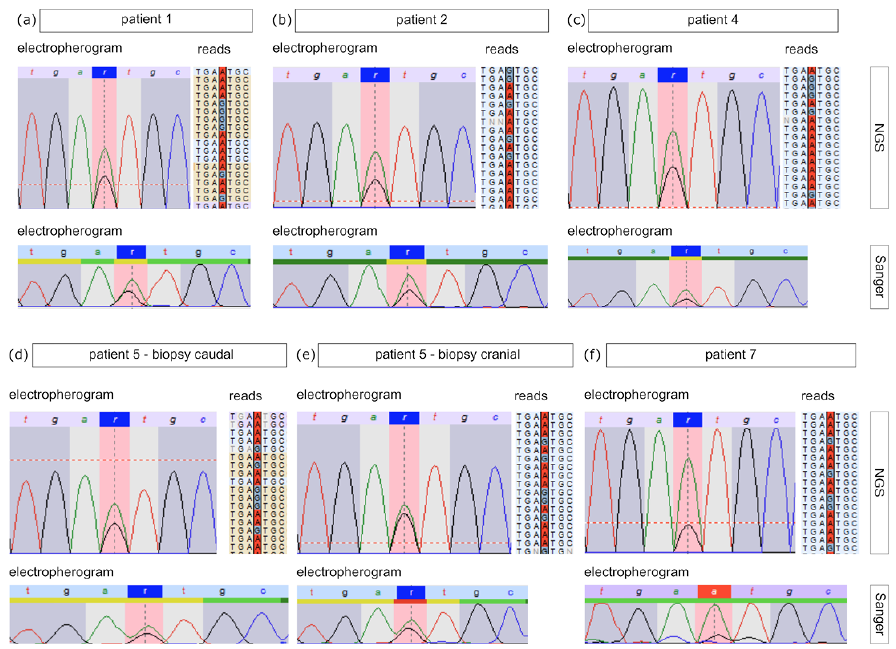 Click to show fullsize
Click to show fullsize