


Hanako Ohmatsu, Makoto Sugaya, Hideki Fujita, Takafumi Kadono and Shinichi Sato
Department of Dermatology, Faculty of Medicine, The University of Tokyo, Tokyo, Japan
Primary cutaneous follicular helper T (TFH)-cell lymphoma has recently been proposed, and is characterized by proliferation of malignant T cells expressing TFH-cell markers, such as CXCL13, accompanied by numerous reactive B cells. We report here a patient whose skin histology showed massive infiltration of both T and B cells, with a proliferation of arborizing high endothelial venules and follicular dendritic cells. Infiltrating T cells were positive for CXCL13, programmed death (PD)-1, inducible T-cell co-stimulator, and BCL-6. Southern blot analyses using DNA from the skin revealed monoclonality of both T and B cells. The patient had marked resistance to treatments, and complete remission was achieved only after allogeneic stem cell transplantation. The present case showed overlapping features with angioimmunoblastic T-cell lymphoma (AITL), although systemic symptoms were not observed. Further study is needed to define the criteria of this provisional entity, representing the cutaneous counterpart of the nodal follicular peripheral T-cell lymphoma or AITL. Key words: follicular helper T; CXCL13; programmed death-1; inducible T cell co-stimulator; angioimmunoblastic T-cell lymphoma.
Accepted Mar 4, 2013; E-pub ahead of print Jun 5, 2013
Acta Derm Venereol 2013; 93: XX–XX.
Makoto Sugaya, Department of Dermatology, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, 113-8655, Japan. E-mail: sugayam-der@h.u-tokyo.ac.jp
Angioimmunoblastic T-cell lymphoma (AITL) is a peripheral T-cell lymphoma characterized by a polymorphous infiltrate, involving lymph nodes with a prominent proliferation of high endothelial venules (HEVs) and follicular dendritic cells (FDCs), termed “angioimmunoblastic pattern”. Clinically, patients with AITL usually present at an advanced stage and show generalized lymphadenopathy, fever, splenomegaly, effusions, arthritis, and skin rashes with polyclonal hypergammaglobulinaemia. The clinical course is aggressive with a median survival of less than 3 years (1). The diagnosis is often difficult, essentially relying on morphological features, including a polymorphous infiltrate with eosinophils and plasma cells, scattered large B-cell immunoblasts, and atypical (sometimes rare) T-cells, often with clear cytoplasm, as well as expansion of FDCs and abundant arborizing HEVs. Epstein-Barr (EB) virus encoded small RNA (EBER)-positive cells are almost always detected (2). It has been reported that tumour cells of AITL express CXCL13, programmed death (PD)-1, inducible T-cell co-stimulator (ICOS), BCL-6, and CD10, markers for follicular helper T (TFH) cells (3–6), which is helpful for diagnosis. However, many cases do not display the whole array of morphological characteristics of AITL, and are classified as borderline cases between AITL and peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS). Monoclonal rearranged bands for T-cell receptor (TCR) genes are detected in most cases. Furthermore, monoclonal rearranged bands for immunoglobulin (Ig) genes are found in some cases, suggesting secondary B-cell lymphoma (7). Cutaneous lesions are present in approximately half of cases with AITL. The skin manifestation of AITL varies widely, such as erythema, papule, bulla and purpura (8, 9). Cutaneous involvement is often related to a monoclonal T-cell proliferation in AITL, even when clinical and histological features are non-specific. CXCL13+ lymphocytes infiltrate the skin of most patients with AITL, and CXCL13 positivity correlates with the presence of a monoclonal T-cell clone (10). These results indicate that CXCL13 is a specific marker not only for systemic AITL, but also for skin involvements of AITL. We describe here the recent case of a patient who presented with primary cutaneous lymphoma, whose skin histology showed characteristics of AITL. The patient showed neither nodal involvement of lymphoma nor clinical symptoms such as fever, splenomegaly, and hypergammaglobulinaemia at the onset of the disease. “Primary cutaneous ATIL” has not been proposed as a new entity, therefore we have to include this case in PTCL, NOS. However, several cases with “primary cutaneous TFH-cell lymphoma” or “mycosis fungoides expressing TFH-cell markers” have been reported recently (11–13). Immunohistochemical study has suggested that our case should fit into this category.
CASE REPORT
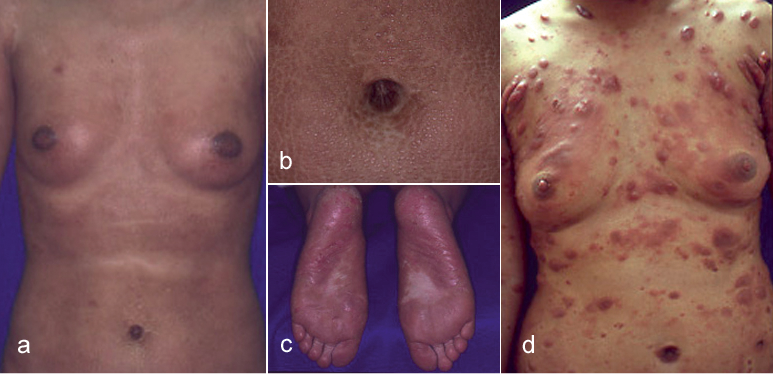
A 26-year-old Japanese woman presented to our department with erythematous to violaceous papules and indurated plaques on her whole body that she had first noticed 4 years earlier (Fig. 1a, b). Thick hyperkeratotic violaceous plaques with erosions were seen on her palms and soles (Fig. 1c). Her superficial lymph nodes were swollen, but she did not show any B symptoms. Computed tomography showed no involvement of deep lymph nodes or internal organs. Laboratory investigations showed a normal full blood count without atypical lymphocytes. The serum level of immunoglobulin E (IgE) was 3,900 IU/ml (normal 0–170 IU/ml) and that of soluble interleukin (IL)-2 receptor was 1,046 U/ml (normal 167–497 U/ml). Serologies for human T-cell lymphotropic virus type I and human immunodeficiency virus were negative. Serological tests for antibodies against EB virus indicated past infection. There were neither autoantibodies detected nor abnormality in serum electrophoresis profiles.
Fig. 1. (a–c) Clinical photographs at the patient’s first visit. (a) Erythematous to violaceous papules and indurated plaques on the whole body. (b) Violaceous indurated plaques in the periumbilical region. (c) Thick hyperkeratotic violaceous plaques on the soles. (d) Many erythematous tumours on the body after 3 years from the first visit.
A biopsy specimen from the abdominal lesion showed mild epidermotropism and dense infiltration of small- to medium-sized lymphocytes in the whole dermis (Fig. 2a, b). The majority of lymphocytes were CD3+ (Fig. 2c), while some lymphocytes in the deep dermis were CD20+ (Fig. 2d). The numbers of CD4+ and CD8+ lymphocytes were almost the same, although the former was slightly more numerous than the latter (data not shown). No CD30+ cells or EBER+ cells were detected. Biopsy specimen of an inguinal lymph node showed no evidence of involvement by lymphoma. Monoclonal TCRγ chain gene rearrangements, but not clonal Ig heavy chain gene rearrangements, were detected using DNA extracts from the skin lesion. Monoclonality of neither T cells nor B cells was detected in the lymph node. The patient was diagnosed with PTCL, NOS. Despite topical steroid therapy, psoralen plus ultraviolet A (PUVA), and administration of interferon (IFN)-γ, she continued to develop plaques and nodules. After 3 years from her first visit, the patient developed many erythematous tumours on the body (Fig. 1d). In addition, oral and intestinal tumours, hepatosplenomegaly, renal and pancreatic nodules, and multiple lymph node swelling appeared. At that time, skin biopsy from her thigh showed diffuse dermal infiltration of lymphocytes without epidermotropism (Fig. 2e). Medium-sized atypical lymphocytes with clear cytoplasm and large atypical lymphocytes infiltrated (Fig. 2f). The proportion of CD20+ lymphocytes was greatly increased compared with that of the first biopsy (Fig. 2g, h). Almost all CD3+ T lymphocytes were CD4+ (data not shown). Both monoclonal TCRγ chain gene rearrangements and monoclonal Ig heavy chain gene rearrangements were detected using DNA extracts from the skin lesion. EBER+ cells were scattered. Oral and intestinal lesions showed diffuse infiltration of T cells and B cells. Two cycles of CHOP therapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) had little effect. Hepatosplenomegaly, intestinal tumours, renal and pancreatic tumours responded to R-hyper CVAD/MA therapy (rituximab, cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate, and cytarabine), but skin lesions and oral tumours were resistant. Four years after her first visit, she received bone marrow transplantation from an unrelated donor after salvage therapy. Her skin and oral lesions went into remission following transplantation. The patient has now been in complete remission for 76 months.
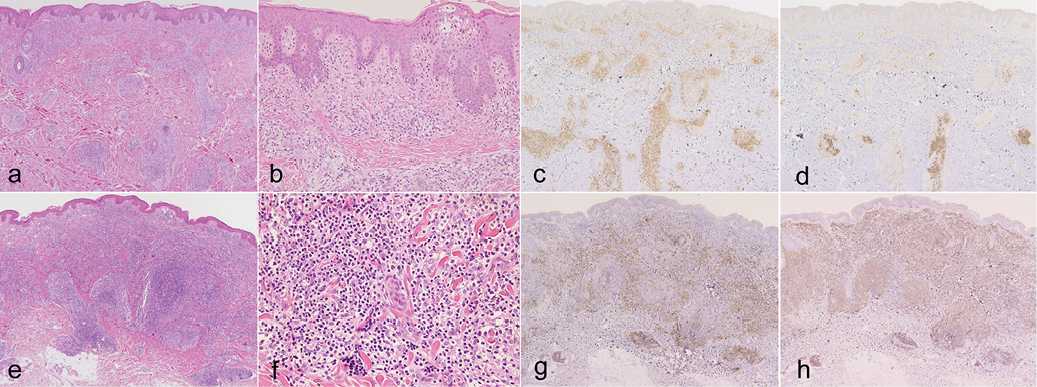
Fig. 2. Histopathology of: (a–d) the first, and (e–h) second skin biopsies. (a, e) Dense lymphocytic infiltration in the whole dermis (haematoxylin and eosin (H&E); original magnification: ×40). (b) Small atypical cells showing epidermotropism (H&E ×200). (c, g) Immunohistochemical staining for CD3 (×40). (d, h) Immunohistochemical staining for CD20 (×40). (f) Medium-sized atypical lymphocytes with clear cytoplasm and large atypical lymphocytes (H&E ×400). (c, d, g, h) Positive cells are stained brown.
Immunohistochemistry
Immunohistochemical study revealed that CD3+ tumour cells were positive for CXCL13 (Fig. 3a), PD-1 (Fig. 3b), and ICOS (Fig. 3c), but negative for CD10. CXCL13 was positive in almost all tumour cells, while PD-1 and ICOS were expressed by approximately half of them. A few tumour cells showed nuclear staining for BCL-6 (Fig. 3d). Some vessels with cubic endothelial cells were morphologically similar to HEVs and positive for PNAd (Fig. 3e). Around PNAd+ venules, CD21+ FDCs were detected (Fig. 3f). These findings were similar to histological characters of lymph nodes in AITL.
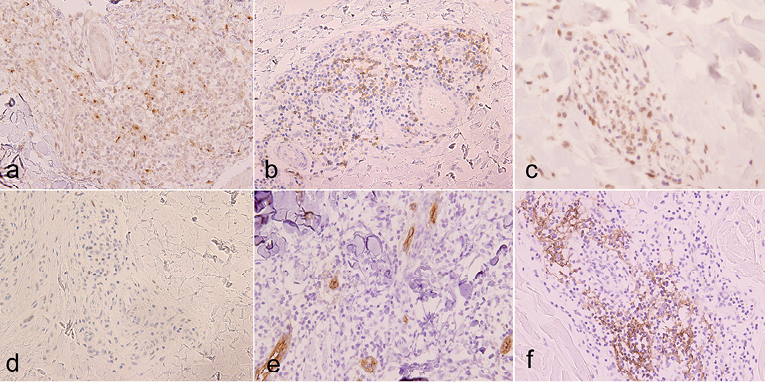
Fig. 3. Immunohistochemical staining for (a) CXCL13, (b) PD-1, (c) inducible T-cell co-stimulator (ICOS) and (d) BCL-6 (×400). Vessels with cubic endothelial cells positive for PNAd (×400) (e). Follicular dendritic cells positive for CD21 (×400). (f) Positive cells are stained brown.
DISCUSSION
The present case showed some features of AITL, such as malignant proliferation of T cells with clear cytoplasm, accompanied by numerous B cells and expansion of FDCs and abundant arborizing HEVs. Clinical presentation, however, was different from AITL, in that our case lacked systemic symptoms, effusions, arthritis, and nodal involvement of lymphoma when skin lesions first developed. It is difficult to diagnose our case as having AITL, systemic proliferation of TFH cells (14).
A subset of nodal PTCL with follicular growth (follicular PTCL), derived from TFH cells, has been identified among PTCL, NOS (5, 15, 16). These lymphomas express TFH-cell markers, such as CXCL13, PD-1, ICOS, BCL-6, and CD10, and may show overlapping features with AITL. The continuity between follicular PTCL and AITL was confirmed by clinicopathological analyses (17, 18). Primary cutaneous TFH-cell lymphoma has been proposed recently (11). Most reported patients had multiple erythematous to violaceous papules, plaques, and nodules, which were more-or-less infiltrated. Notably, B symptoms were absent. Patients with diffuse disease showed marked resistance to treatments, with only one case of complete remission after allogeneic hematopoietic stem cell transplantation. Other researchers reported similar cases as “mycosis fungoides expressing TFH-cell markers” (12, 13). We believe that our case fits into this category, although our case showed epidermotropism (Fig. 2b). Moreover, tumour cells in our case did not express CD10, one of the most widely used TFH-cell markers. We, however, did not detect CD10 expression in skin lesions of AITL (data not shown). Consistent with our findings, CD10 has been reported to be less sensitive than PD-1 or CXCL13, both in skin and lymph nodes of AITL (5, 10, 16, 17). Other types of cutaneous T-cell lymphoma, such as primary cutaneous CD4+ small/medium T-cell lymphoma, were reported to express TFH-cell markers, such as CXCL13, whose clinical presentation is different from our case (19). Overall clinicopathological presentation supported that our case was primary cutaneous TFH-cell lymphoma.
We detected proliferation of PNAd+ HEVs in the lesional skin, which is one of the characteristic findings in AITL. However, it was previously reported that approximately two-thirds of inflammatory skin lesions and cutaneous lymphoma had PNAd+ venules, while normal skin venules did not express PNAd (20). Vascular PNAd expression correlated with the extent of mononuclear cell infiltration. Moreover, many of these venules showed morphological features identical to those of HEV in lymphoid tissues. We also confirmed that most cases of skin involvement by AITL and mycosis fungoides contained PNAd+ venules (data not shown). Therefore, vascular PANd expression may play a role in the dermal infiltration of lymphocytes, although this not a finding specific to cutaneous lymphoma.
In summary, we report here a case of primary cutaneous TFH-cell lymphoma that was treated with allogeneic bone marrow transplantation. This case showed overlapping features with AITL, although systemic symptoms and nodal involvement were not observed. Further research is needed to define clearly the criteria of this provisional entity, representing the cutaneous counterpart of nodal follicular PTCL or AITL.
REFERENCES

