


Departments of Dermatology, 1Federal and Catholic University of Pelotas, 96075-320 Pelotas, Brazil, 2University of Freiburg, Freiburg, Germany, 3Catholic University of Porto Alegre, and 4Electron Microscopy, EMBRAPA-CPA-CT Pelotas, Pelotas, Brazil. E-mail: hiramalmeidajr@hotmail.com
Accepted May 30, 2016; Epub ahead of print Jun 8, 2016
MEND syndrome (male emopamil-binding-protein disorder with neurological defects) is a syndromic ichthyosis with neurological anomalies associated with emopamil-binding protein mutations. This denomination was proposed in 2012 (1). It is an X-linked condition with no mosaicism, unlike Conradi-Hünermann-Happle (CHH) syndrome, which is X-linked dominant and usually lethal in males. Some male cases of CHH syndrome have survived, always associated with mosaicism, reflecting either XXY constitution or a post-zygotic new mutation (2).
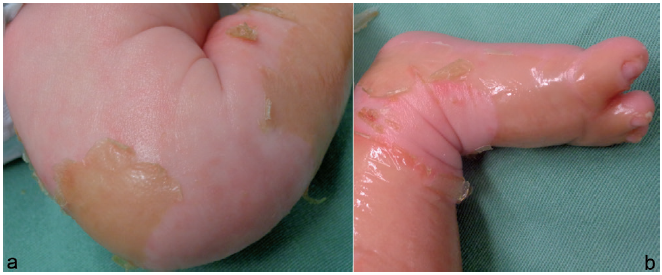
MEND syndrome presents as collodion baby, with distinct facial features, such as a prominent nasal bridge, low-set ears and large anterior fontanelle. Neurological defects include cerebellar hypoplasia, hypoplasia of corpus callosum, hydrocephalus, hypotonia, development delay and seizures. Polydactyly and syndactyly are also present, characterizing this syndromic ichthyosis (1).
MEND syndrome is caused by mutations in the gene encoding the emopamil-binding protein (EBP), located on the short arm of the X chromosome. EBP is a sterol isomerase responsible for one of the final steps in the production of cholesterol. Inhibition of cholesterol biosynthesis leads to the accumulation of sterol precursors and low concentration of intracellular cholesterol, although with normal serum cholesterol level. In cases of CHH syndrome, the high doses of cholesterol precursors are not directly associated with the severity of the phenotype (3, 4).
A 2-week-old male patient examined in the neonatal intensive care unit (ICU) presented with a yellowish collodion membrane involving the trunk and limbs (Fig. 1, and Fig. S1a). Dehydration and gradual peeling of this membrane had occurred over a period of 2 weeks. Polydactyly and syndactyly became more evident after elimination of the membrane. The shed membrane was collected for scanning electron microscopy (SEM), both after fixation and in the dry state.
Fig. 1. Clinical characteristics. (a) Thigh and right leg partially covered by collodion membrane. (b) Right foot and leg showing collodion membrane.
On clinical examination, his mother presented atrophic hypopigmented lesions on the abdomen (Fig. S1b) and a linear hyperpigmented lesion on the right arm. Ophthalmological examination showed focal cataract. The patient’s grandmother reported that 30 years ago she gave birth to a male who died in the first days of life, presenting skin changes (“burned skin”) and syndactyly.
The patient died of respiratory complications at the age of 8 weeks.
Pathological calcifications in bone epiphyses were not visible on X-rays; however, calcifications were observed in the soft tissue. Computed tomography (CT) scanning of the skull showed agenesis of corpus callosum, cerebellar hypoplasia, agenesis of cerebellar vermis and Dandy-Walker cyst. Microphthalmia was also present (Fig. S2a). DNA was isolated from peripheral blood and sequenced for the EBP gene in both the newborn and his parents. In exon 4 the mutation c. 439C > T, p. R147C was found in both the newborn and his mother (Fig. S2b). This amino acid substitution is predicted to be “probably damaging” (PolyPhen2) and “disease causing” (Mutation Taster, http://www.mutationtaster.org/), respectively.
Analysis of the collodion membrane was performed using SEM. The external surface showed intense desquamation of corneocytes. Perforating hair follicles with corneocytes accompanying the hair growth were also observed (Fig. 2), differently from normal controls’ epidermal surface, which showed a less intense desquamation (Fig. S3a). The internal surface revealed the presence of nucleated corneocytes, possibly characterizing a parakeratotic differentiation of the collodion membrane (Fig. S3b). Laterally, multiple layers of corneocytes were seen, characterizing a thick membrane (Fig. S3c).
Fig. 2. Scanning electron microscopy (SEM). (a) External surface of the collodion membrane with intense desquamation of corneocytes and visible projections of the hair follicles (× 150).
MEND syndrome has been defined recently as a separate entity from CHH syndrome (1).
The present case showed several phenotypic characteristics consistent with previous publications reporting keratinization disturbance, such as a collodion membrane, polydactyly, syndactyly and microphthalmia, the central nervous system (CNS) findings also corroborated with previous findings (1). A recent report expanded the MEND phenotype, with a family with 4 affected males, who were not born with collodion membrane and showed behavioural difficulties without ocular or skeletal abnormalities (5).
The SEM findings revealed a thick collodion membrane with intense desquamation of corneocytes on the external surface, differently from normal controls. In the internal surface nuclei were seen in the lower corneocytes, characterizing a parakeratotic collodion membrane. There are no reports of SEM findings for collodion membrane in MEND syndrome to compare with our results.
There is a report of SEM findings for a collodion membrane of a self-healing collodion baby, which showed a compact membrane at the external surface, without corneocyte elimination (6). In this report, examination of the inner surface did not identify nuclei in the lower corneocytes (6); these findings need to be compared with further cases of collodion babies. It is very likely that ultrastructural aspects of collodion membranes are age-related and differ corresponding to the respective molecular defects.
DNA sequencing of the EBP gene of the current patient showed the mutation, p.R147C, which is capable of causing this condition. In the same codon, the mutations p.R147H and p.R147G have been reported previously in the literature in patients with CHH syndrome (7–9). Two reported EBP mutations leading to amino acid substitutions, p.W47C and p.L18P, such as the one described here, were associated with MEND syndrome (10, 11). They are compatible with life in males (12); however, the phenotype remains very severe. In the current report, the patient’s mother showed oligosymptomatic changes and it is very likely that her mother carried also an EBP mutation, since she reported the loss of a male newborn with cutaneous and skeletal involvement. Careful clinical examination, for identifying such cases, and genetic counselling are essential.
Although MEND is rare, new cases need to be reported to better understand and recognize this disorder, and therefore, to expand knowledge to establish genotype-phenotypic correlations, including ultrastructural aspects.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize